An Imposter Twice Over: A Case of IgG4-Related Disease





Immunoglobulin G4-related disease (IgG4-RD) is an immune-mediated fibroinflammatory condition that involves multiple organs and appears as syndromes that were once thought to be unrelated. This disease leads to mass lesions, fibrosis, and subsequent organ failure if allowed to progress untreated.1 Involvement of gastrointestinal (GI) organs, salivary glands, lacrimal glands, lymph, prostate, pulmonary, and vascular system have all been reported.2 Elevated IgG4 serum levels are common, but about one-third of patients with biopsy-proven IgG4-RD do not manifest this characteristic.3,4
Diagnostic confirmation is with biopsy, and all patients with symptomatic, active IgG4-RD require treatment. Glucocorticoids are first-line treatment and are utilized for relapse of symptoms. In addition to glucocorticoids, steroid-sparing medications, including rituximab, azathioprine, mycophenolate mofetil, tacrolimus, and cyclophosphamide have all been used with successful remission.5,6 Here, the authors discuss a case of IgG4-RD that presented with intrahepatic biliary obstruction (mimicking cholangiocarcinoma) and subsequent development of coronary arteritis despite treatment.
Case Presentation
In June 2015, a 57-year-old Air Force veteran presented to Eglin AFB Hospital with pruritic jaundice and acute abdominal pain. He was found to have elevated bilirubin levels (total bilirubin 10 mg/dL [normal range 0.2-1.3 mg/dL], direct bilirubin 6.6 mg/dL [normal range 0.1-0.4 mg/dL]). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) also were moderately elevated (147 U/L and 337 U/L, respectively). 
Prior to this presentation, the patient had been in his usual state of health. His past medical history was notable only for minimal change kidney disease (MCD). MCD is defined as effacement of the podocyte seen on electron microscopy, which allows the passage of large amounts of protein. 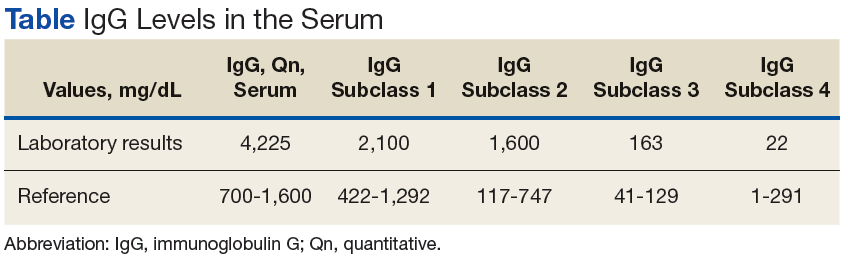
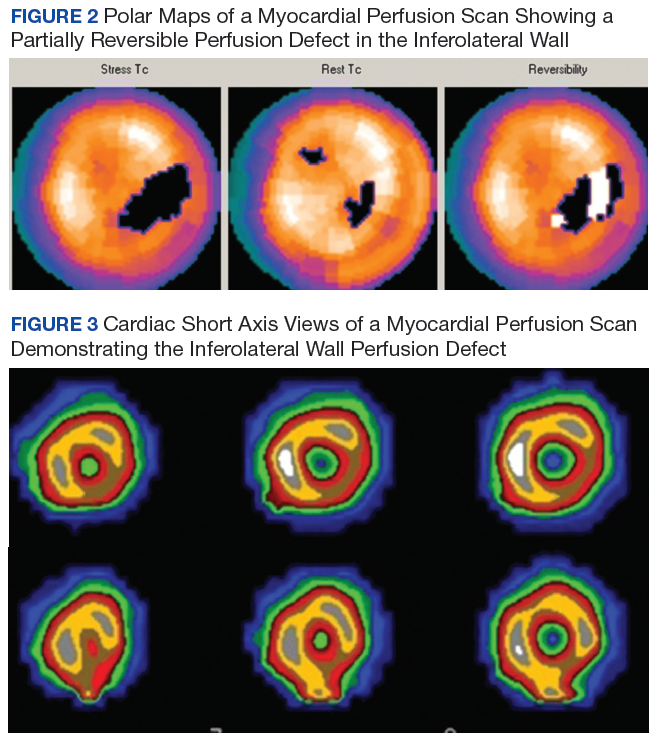
A cholangiogram showed abnormal filling into the left main intrahepatic duct and obvious obstruction at the bifurcation of the bile duct. A biliary drainage catheter was placed, and a repeat cholangiogram 2 days later showed involvement of both right and left intrahepatic ducts. The distal common bile duct appeared uninvolved as did the pancreas. Lymphadenopathy was noted at the liver hilum. Klatskin cholangiocarcinoma (type IIIB) was the presumed diagnosis. Based on these findings, tumor resection was performed 3 weeks later, including left hepatectomy, caudate lobe resection, complete bile duct resection, cholecystectomy, with reconstruction by Roux-en-Y intrahepaticojejunostomy. In addition, portal and hepatic artery lymph node dissection was completed.
Surgical specimens were sent for pathologic evaluation and were found negative for malignancy. Patchy areas of storiform fibrosis, obliterative phlebitis, and lymphoplasmacytic infiltrate were noted. IgG4 immunostain highlighted the presence of IgG4 positive plasma cells with a peak count of 145 IgG4 positive plasma cells/hpf. About 80% of the plasma cells were positive for IgG4. Unusually dense eosinophilic infiltrate with plasma cells and regions of dense fibrosis that strongly contributed to the masslike appearance on CT imaging also were noted. Final histology confirmed the diagnosis of IgG4-RD. Elevated levels of total IgG in the serum were observed without elevation in serum IgG4 (Table).
The patient was started on prednisone 40 mg and azathioprine 150 mg daily, with subsequent taper of prednisone over the next 6 months. After prednisone was discontinued, the patient reported new symptoms of lower extremity pain, neuropathy, and swelling of his face. Laboratory results were notable for elevated erythrocyte sedimentation rate. The patient was restarted on prednisone 40 mg daily. Azathioprine was replaced with a regimen of 4 doses every 6 months of IV rituximab 700 mg q week and mycophenolate mofetil (1,000 mg bid). After remission was induced, the patient was slowly weaned off prednisone again.
Following 6 months of successful discontinuation of prednisone and continued rituximab and mycophenolate mofetil therapy, the patient presented to the emergency department with new onset chest pain and shortness of breath. A CT angiography of the chest showed right upper and middle lobe infiltrate, and he was treated for community acquired pneumonia. Additionally, he was noted to have elevated troponin levels suggestive of myocardial infarction (MI). Initial troponin was 1.23 ng/mL (normal range < 0.015 ng/mL), which trended down over the next 18 hours. A bedside echocardiogram showed a normal left ventricular ejection fraction without wall motion abnormalities. Etiology for his acute MI was presumed to be demand ischemia from fixed atherosclerotic plaque. Further inpatient cardiac risk stratification was changed to the outpatient setting, and he was started on medical management for coronary artery disease with a beta blocker, a statin, and aspirin. He was discharged home on 10 mg prednisone daily, which was subsequently tapered over several weeks.
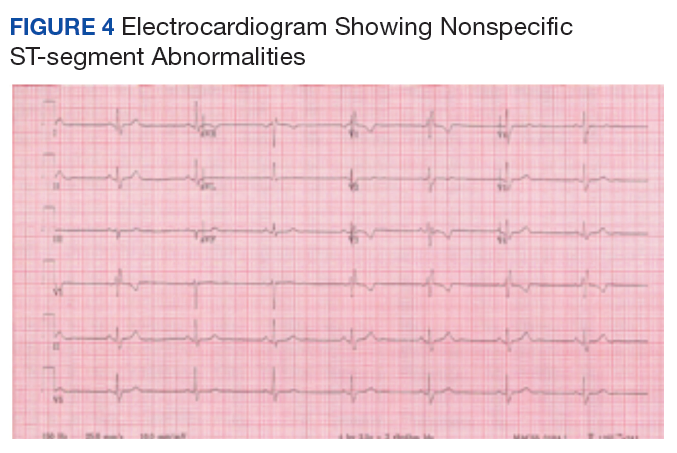
In follow-up, a Lexiscan myocardial perfusion imaging was conducted that demonstrated an inferolateral defect and associated wall motion abnormalities (Figures 2 and 3).