A Veteran Presenting With Chronic Progressive Dyspnea on Exertion





Alternatively, pulmonary arterial hypertension (PAH) is suspected in patients with no identifiable cause of PH, pulmonary artery wedge pressure 15 mm Hg and pulmonary vascular resistance of 3.0 Wood units.6 Importantly, PAH is not synonymous with PH but is a circumspect PH disease subgroup. In turn, PAH may be idiopathic, hereditary, or associated with other select, predisposing disorders, namely systemic sclerosis. In PAH, the interplay between genetic and molecular factors results in effacement of distal pulmonary arterioles due to plexigenic, fibrotic, and/or concentric hypertrophic remodeling. Increased vascular resistance promotes early right ventricular dilation and impaired systolic function. As a result, patients with PAH are at particularly elevated risk for cor pulmonale.
►Dr. Clark: Overnight oximetry revealed baseline oxygen saturation of 94%, an oxygen nadir of 84% with a total of 7 minutes with oxygen < 90%. On a 6-minute walk test, the patient had a max heart rate of 116 and oxygen nadir of 93%. Chest CT with and without contrast showed no evidence of pulmonary emboli but noted mild emphysematous changes. A V/Q revealed no evidence of acute or chronic pulmonary thromboembolic disease. Coronary catheterization showed normal coronary anatomy without significant CAD. A right heart catheterization showed findings consistent with severe PH with normal left-sided filling pressures (Table 3).
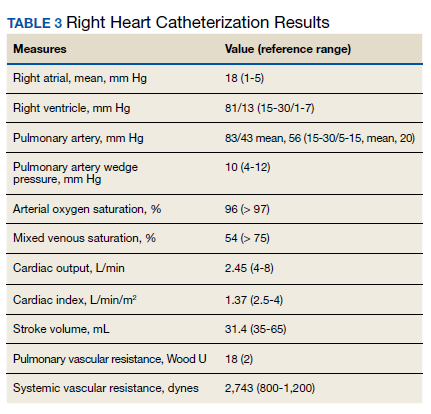
The patient returned a normal antinuclear antibody, C-reactive protein, HIV, and liver function panel. Based on these findings, a presumptive diagnosis of group 1 PH (idiopathic PAH) was made. Given the severity of his right heart dysfunction, he was transferred to the cardiac care unit and initiated on epoprostenol.
Dr. Maron, can you review the different treatment options for idiopathic PAH and explain why epoprostenol was chosen for this patient?
►Dr. Maron: There are 14 US Food and Drug Administration-approved drug therapies for patients with PAH, which all target either nitric oxide signaling, endothelin receptors, or the prostacyclin pathway. In the current era, treatment-naïve patients with PAH are generally initiated on calcium channel antagonist therapy if there is evidence of vasoreactivity during right heart catheterization (following nitric oxide administration), dual therapy most often with an endothelin receptor antagonist and phosphodiesterase inhibitor, or parenteral prostacyclin therapy. Since < 5% of patients will demonstrate vasoreactivity, the decision at point of care in incident patients with PAH often focuses on dual oral therapy or initiation of parenteral prostacyclin therapy. In this case, the patient reported presyncope with minimal physical activity (eg, bending over or walking up stairs) and severely decreased functional status (ie, New York Heart Association Functional [NYHA] Class III – IV), and he had a cardiac index within the range of cardiogenic shock (< 2.0 L/min/m2). Collectively, this clinical profile is considered particularly high risk, therefore, a recommendation for parenteral continuous prostacyclin therapy was made.
► Dr. Clark: The patient tolerated epoprostenol and reported improvement in his symptoms. He had a tunneled line catheter placed for continuous epoprostenol infusion. He was discharged home and scheduled for outpatient follow-up in a PH clinic. At 4 months following discharge, he was reporting steady clinical and functional improvement as well as improvement in his dyspnea. A second therapy (oral phosphodiesterase type-V inhibitor) was initiated and tolerated well. Overall, he reported resolution of presyncope, NYHA Functional Class II symptoms, and the absence of important drug effects.