Incidentally Discovered Ochronosis Explaining Decades of Chronic Pain





Discussion
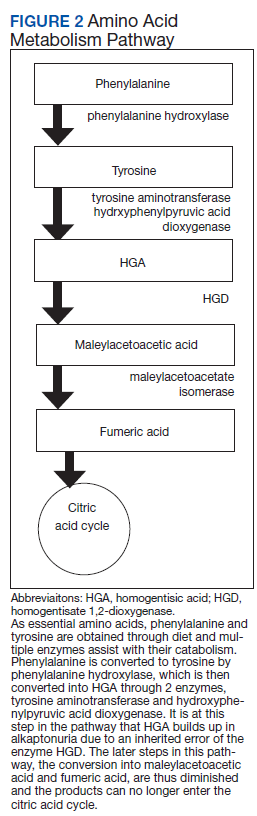
Alkaptonuria is a rare autosomal recessive disorder, with a prevalence of about 1 in 100,000 to 250,000, which results from an enzyme error in an essential amino acid metabolism pathway (Figure 2).1 This inheritable gene mutation leads to ineffective homogentisate 1,2-dioxygenase (HGD), an enzyme required to break down HGA—which is a product of phenylalanine and tyrosine metabolism.2 As these patients engage in normal dietary protein intake, which includes essential amino acid phenylalanine, they develop clinically evident manifestations of the buildup and deposition of HGA.
The rarity of alkaptonuria combined with the gradual buildup of HGA makes it difficult to diagnose. A common diagnostic technique is the visualization of discolored cartilage during surgical procedures, especially when discoloration in urine or skin is not immediately evident. A few case reports have noted surgical diagnosis of black or darkening tissue, known as ochronosis, following tendon rupture—a common complication of this disorder.3-5 Additional intervention-related case reports linked to the discovery of ochronosis include aortic valve replacement, lumbar discectomy, and bronchoscopy.6-9 Cases like these illustrate the complex, disabling, and unclear nature of this disorder when not diagnosed early in life.
The patient in this case communicated via e-mail about his tendon repair surgery. “Something very interesting was found during the surgery,” the patient explained. “I was diagnosed with the disease called ochronosis. I don’t know much about this disease but I am beginning to know why some of the things are happening to me and why I am always in constant pain.” This was the first recognized clue toward a diagnosis of alkaptonuria.
Pathophysiology
The pathophysiology of alkaptonuria is based on the extensive deposition of HGA throughout the body. Its progression is based on 3 clinical stages: clinical silence, clinical ochronosis, and ochronotic arthropathy.1 In the first stage the disorder is asymptomatic but includes its most notable feature—the gradual darkening of urine when exposed to air through oxidation of the renally excreted HGA. A similar process occurs in the blood through formed HGA-melanin compounds, which cause discoloration in cartilage.1 This internal metabolic disruption accounts for the disorder’s eventual second stage, clinical ochronosis, usually with an onset in the second or third decade. Prominent features noted on physical examination primarily include discoloration of ear pinnae and eye sclera but can involve the nose, gums, teeth, and hands. The third, final, and symptomatic stage, ochronotic arthropathy, occurs by the patient’s fourth to fifth decade and presents as joint pain, usually starting with the vertebrae and larger joints like hips, knees, and shoulders, that can appear as advanced early osteoarthritis on imaging.
Treatment
This clinical manifestation of alkaptonuria requires that HCPs manage patients with 3 strategies: decrease HGA buildup, alleviate symptoms, and monitor for disorder complications. Decreasing HGA buildup is a difficult aspect of management given the natural physiology of protein intake and metabolism. Three approaches to limit HGA buildup incorporate decreasing protein intake, inhibiting enzyme production of HGA, and increasing HGA excretion. Phenylalanine is an essential amino acid—meaning its levels are dependent on dietary protein intake. Patients should be advised to adhere to a low protein diet, especially phenylalanine and tyrosine, to lessen HGA concentrations.