Acute Encephalopathy Following Hyperbaric Oxygen Therapy in a Patient on Metronidazole





Discussion
This patient’s encephalopathic symptoms correlate temporally with the onset of HBOT. There is no medical literature suggesting a relationship between HBOT and encephalopathic symptoms with MRI abnormalities, and in fact, some studies suggest HBOT as a treatment for hypoxic-ischemic encephalopathy in neonates.11 This led us to believe that the HBOT may have exacerbated some underlying condition, evidenced by the specific MRI findings of T2 fluid-attenuated inversion recovery (FLAIR) hyperintensities in the dentate nuclei and inferior colliculi (Figures 1 and 2). 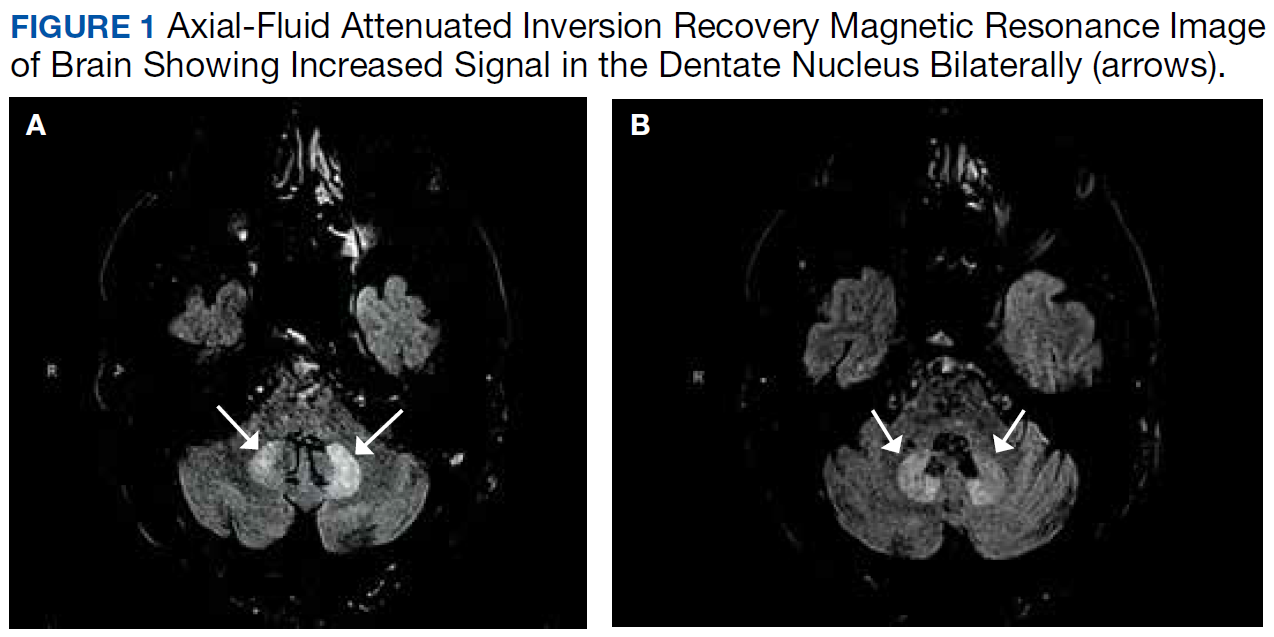
Differential diagnoses for T2 hyperintense lesions in the dentate nuclei include metronidazole toxicity, acute Wernicke encephalopathy (WE), and methyl bromide intoxication. Diseases that would have presented in infancy with similar MRI findings (Canavan disease, maple-syrup urine disease, and glutaric aciduria type 1) were not considered plausible.12-14 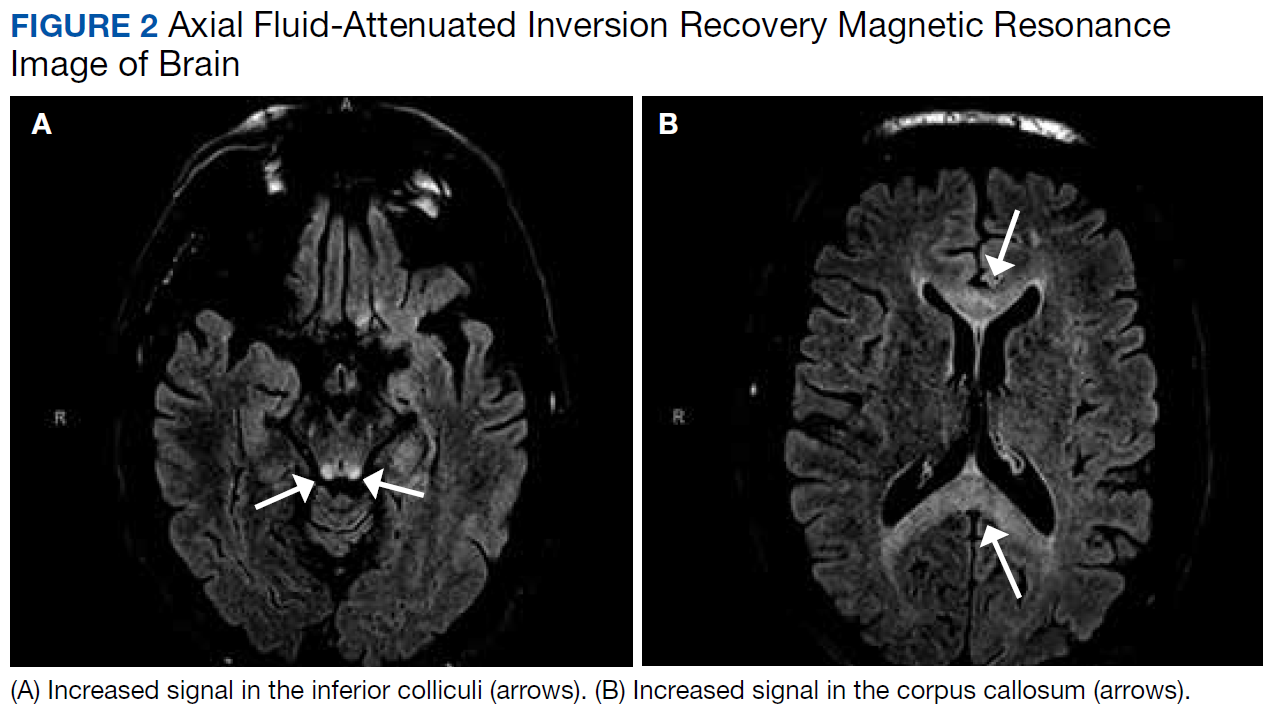
Despite his denial of alcohol use, the patient was at risk for malnutrition secondary to his mandibular lesion and difficulty eating. Clinically, he presented with episodes of confusion and ataxia, consistent with 2 of the classic triad of symptoms of WE (no ocular abnormalities noted on exam). Typical MRI findings in WE include signal intensity alterations (including T2 hyperintensities) in the medial thalami, mammillary bodies, collicular bodies, and periaqueductal and periventricular regions.14,15 Atypical MRI findings in WE include symmetric signal intensity changes in the cerebellum, dentate nuclei, caudate nuclei, red nuclei, cranial nerve nuclei, and splenium.14 Of note, atypical MRI findings were more common in patients without alcohol use disorders and WE, and typical MRI findings were more common in patients with alcohol use disorders.14 However, this patient’s report of no alcohol use and the serum thiamine level being within normal limits (173 nmol/L; range 78-185 nmol/L) made acute WE less likely than metronidonazale-induced encephalopathy (MIE).
The most common neurologic AE of metronidazole is distal symmetric sensory polyneuropathy, which also can have motor or autonomic features.16,17 While our patient had a history of peripheral neuropathy, he noted marked worsening of foot pain 3 months after initiating metronidazole therapy. A potential mechanism involves metronidazole or its cytotoxic intermediates binding neuronal ribonucleic acids, thus inhibiting protein synthesis and resulting in degenerative neuronal changes and reversible axonal swelling (as opposed to the DNA interference attributed to the drug’s mechanism of bactericidal action).18 Neuropathies may result from prolonged high-dose metronidazole therapy (cumulative dose > 42 g),3 but they also have been seen in short-term use of high dosages.17
CNS AEs are much rarer and are thought to be associated with metronidazole’s ability to cross the blood-brain barrier. These patients present as a toxic encephalopathy with cerebellar dysfunction (dysarthria, ataxia) as the most common presentation, followed by AMS and seizures.4 Our patient presented with acute confusion and ataxia. Animal studies suggest that γ-aminobutyric acid (GABA) receptor modulation in the cerebellar and vestibular systems may contribute to this neurotoxicity, but no definitive mechanism of injury has been found.19
On MRI, MIE most commonly presents with hyperintense lesions in the bilateral cerebellar dentate nucleus on T2-weighted and FLAIR images.5,20 The midbrain, dorsal pons, medulla, and corpus callosum also can show increased signal intensity.5 This AE does not seem to be dose- or duration-dependent, and most cases report complete or partial resolution of symptoms following discontinuation of the drug, though this is not absolute.4,13,21 The patient’s MRI findings were highly consistent with MIE (Figure 2).