Unsuspected Lymphomatoid Granulomatosis in a Patient With Antisynthetase Syndrome





Clinical diagnosis of lymphomatoid granulomatosis (LYG) often is difficult, especially in patients with multiple comorbidities. We present a 60-year-old woman with worsening fatigue, night sweats, unintentional weight loss, and dyspnea of 2 weeks’ duration. Her medical history was remarkable for recent radiation therapy for recurrent breast cancer and antisynthetase syndrome complicated by interstitial lung disease and controlled with azathioprine. Computed tomography showed ground-glass opacities and nodular infiltrates in all lung lobes, raising concern for radiation pneumonitis and drug toxicity. Skin examination revealed erythematous and hemorrhagic papules, macules, and blisters on the lower leg. A diagnosis of LYG was made on a skin biopsy showing large angiocentric Epstein-Barr virus (EBV)–positive B cells. The patient soon progressed to develop nodal large B-cell lymphoma and died 6 weeks later. This rare report of LYG in a patient with antisynthetase syndrome highlights the diagnostic difficulty and aggressive course of LYG as well as the important role of skin biopsy in establishing the diagnosis.
Practice Points
- Lymphomatoid granulomatosis (LYG) is a rare extranodal angiocentric large B-cell lymphoma driven by the Epstein-Barr virus.
- Lymphomatoid granulomatosis should be suspected when immunocompromised patients present with nodular lung infiltrates and/or nonspecific skin lesions.
- Skin biopsy serves a critical role in establishing the diagnosis of LYG, especially when clinical and radiologic findings are obscured by other comorbidities.
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–related extranodal angiocentric lymphoproliferative disorder. Most patients are adults in the fifth decade of life, and men are twice as likely as women to be affected.1 The most common site of involvement is the lungs, which has been observed in more than 90% of patients.2 The skin is the most common extrapulmonary site of involvement with variable manifestations including “rash,” subcutaneous nodules, and ulceration. Although a small subset of patients experience remission without treatment, most patients report a progressive course with median survival of less than 2 years.1,2 Clinical diagnosis often is challenging due to underrecognition of this rare condition by multidisciplinary physicians.
Case Report
A 60-year-old woman presented with fatigue, night sweats, poor appetite, unintentional weight loss, and dyspnea with minor exertion of 2 weeks’ duration. Her medical history was remarkable for antisynthetase syndrome manifested as polymyositis and interstitial lung disease, as well as recurrent breast cancer treated with wide excision, chemotherapy, and radiation therapy completed 2 months prior. Antisynthetase syndrome was controlled with azathioprine for 2 years, which was stopped during chemotherapy but restarted to treat worsened myalgia 4 months prior to presentation. Two weeks prior to hospital admission, she was treated with antibiotics at an outside hospital for presumed pneumonia without improvement. Upon admission to our hospital she was pancytopenic. Chest computed tomography showed interval development of extensive patchy ground-glass opacities in all lung lobes with areas of confluent consolidation. Broad infectious workup was negative. Given the time course of presentation and anterior accentuation of the lung infiltrates, the greatest clinical concern was radiation pneumonitis followed by drug toxicity. A bone marrow biopsy was hypocellular but without evidence of malignancy. Her pancytopenia was thought to be induced by azathioprine and/or antibiotics. Antibiotics were discontinued and prednisone was started for treatment of presumed radiation pneumonitis.
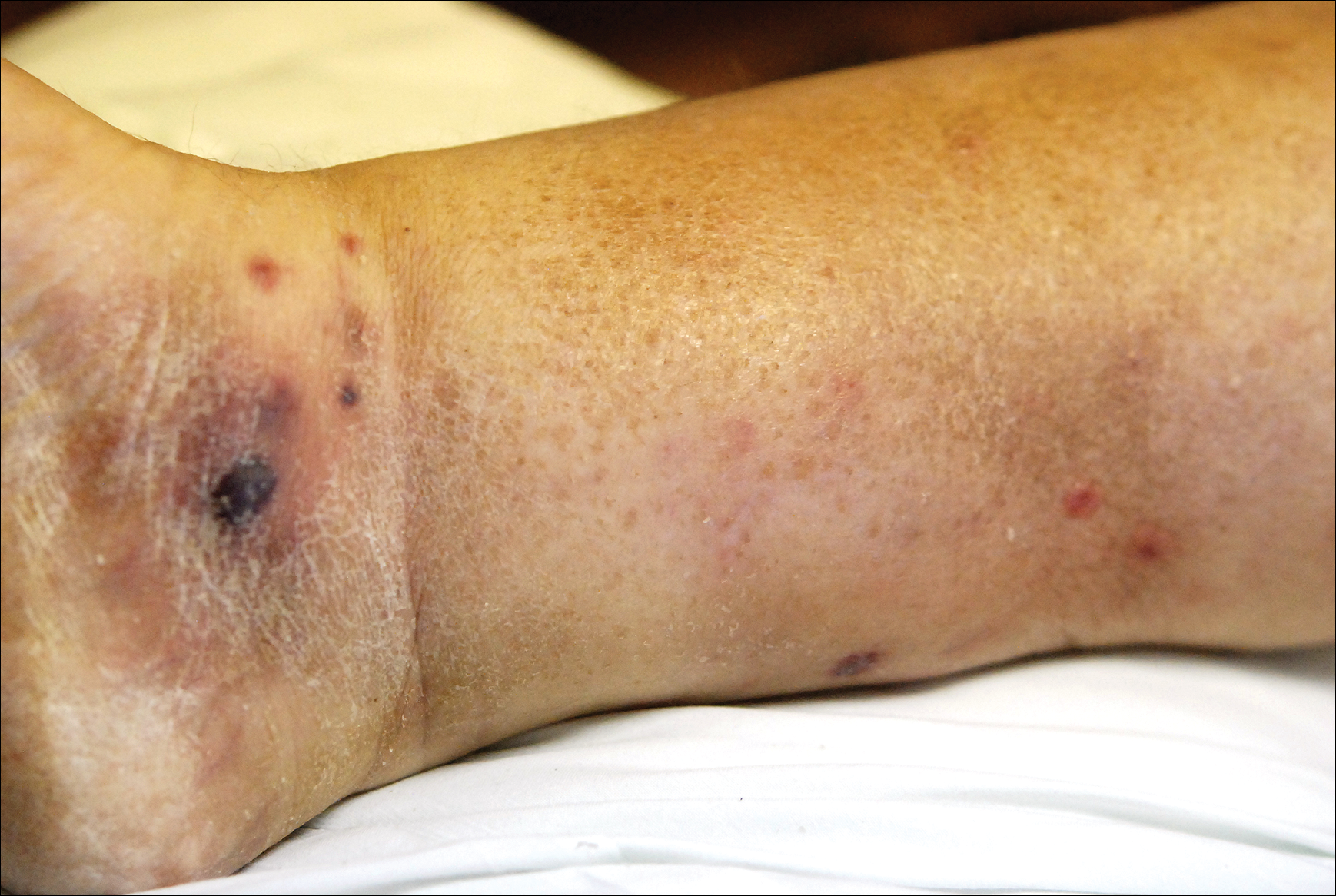
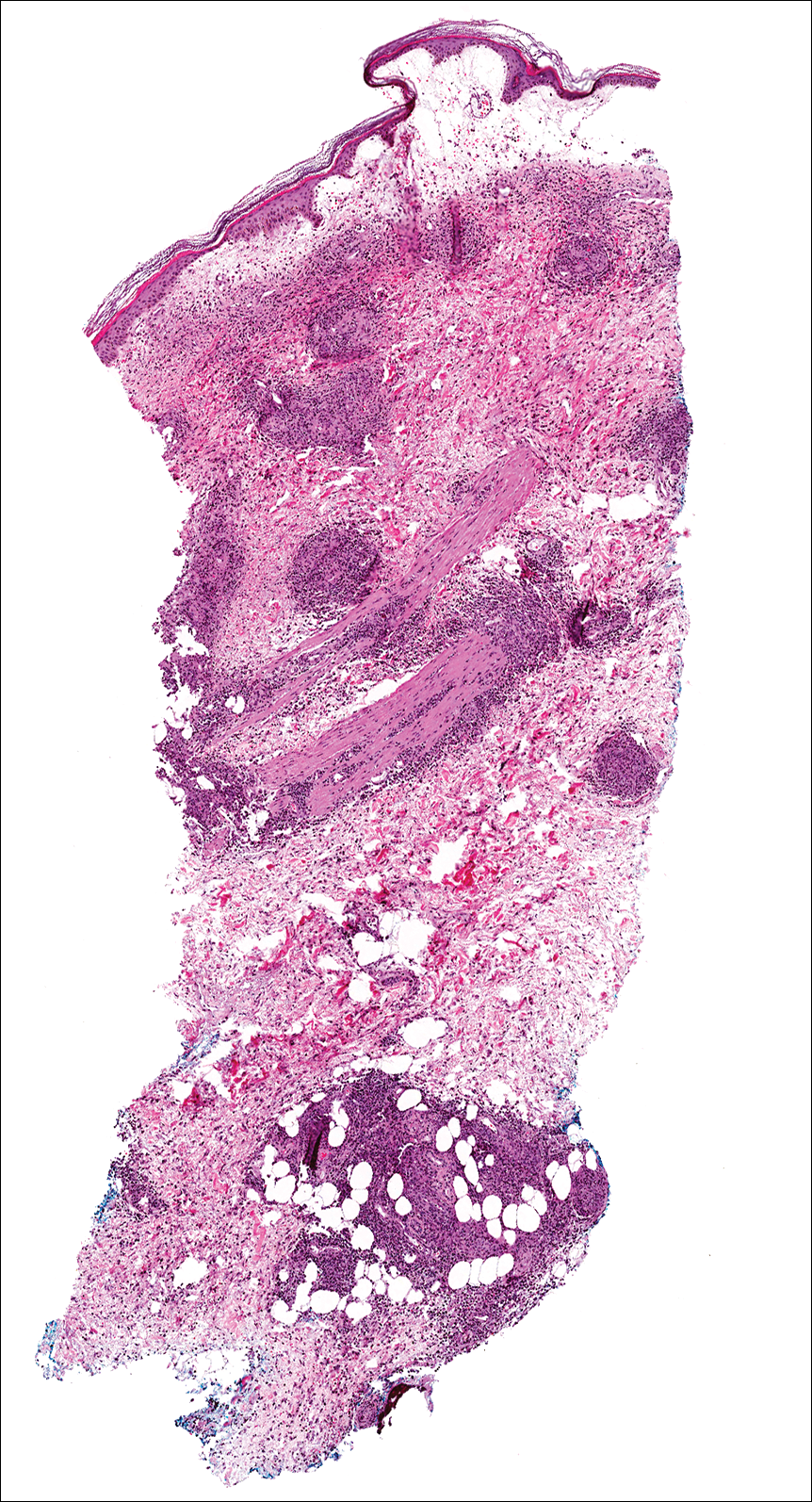
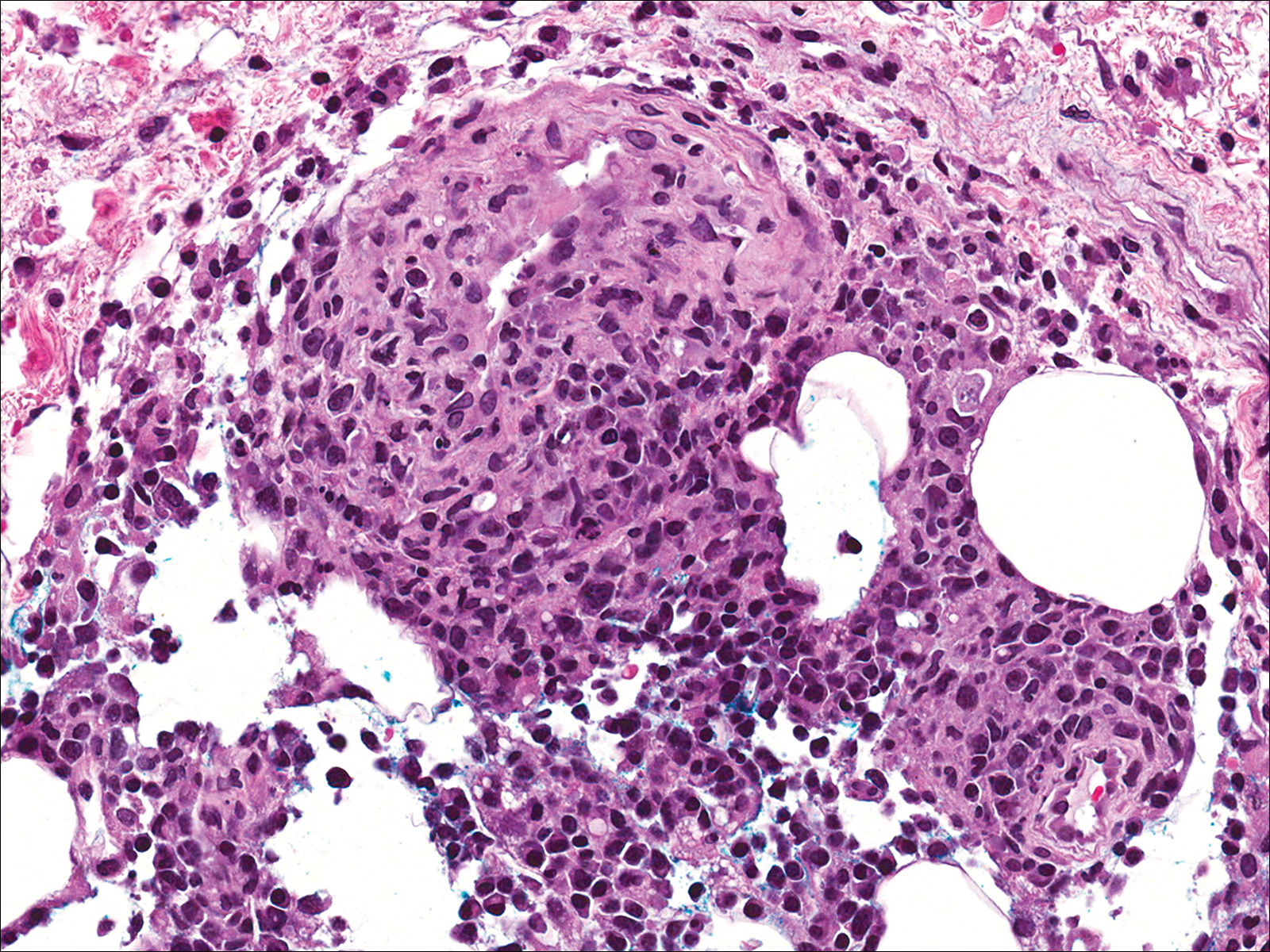
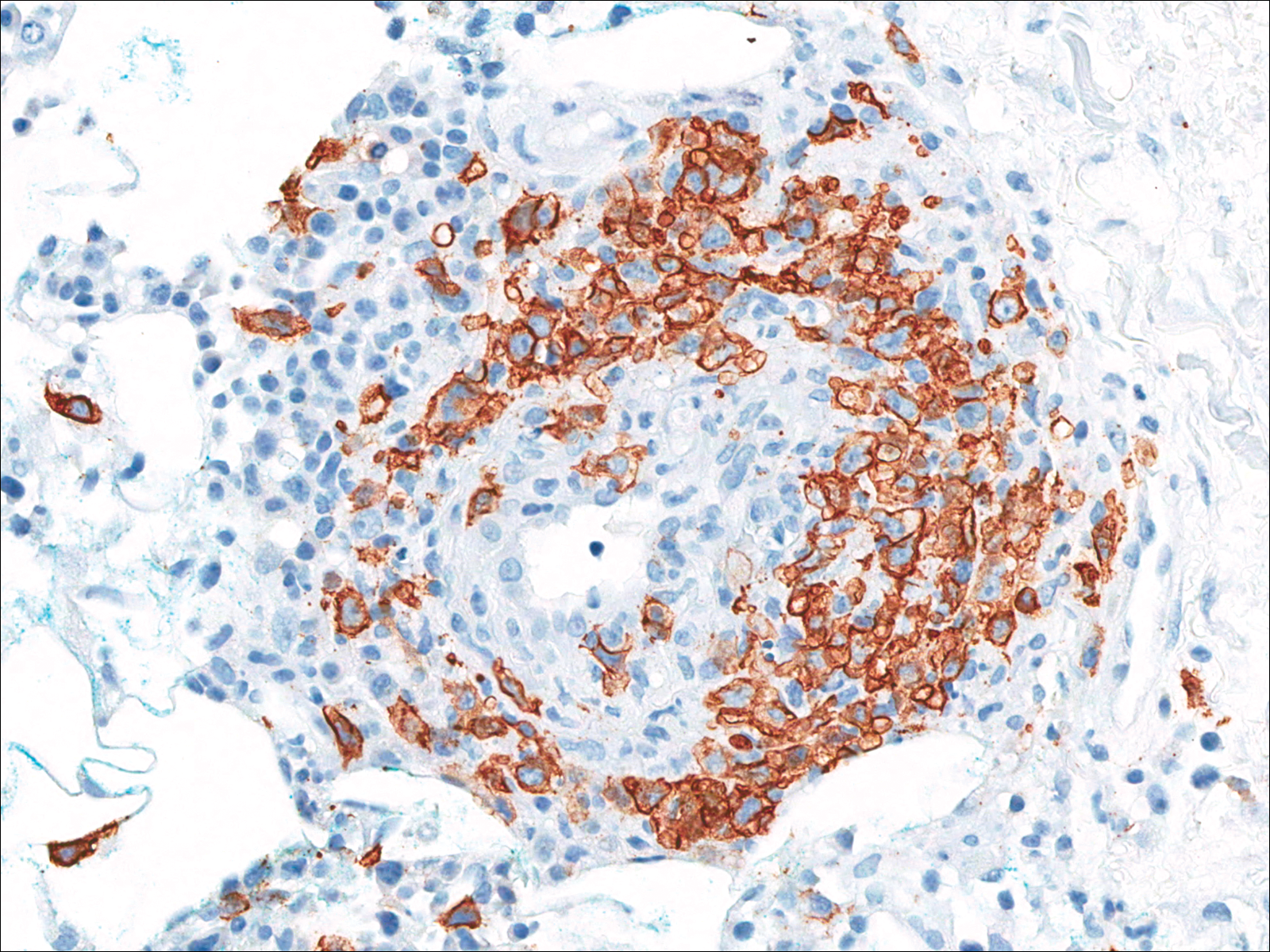
A few days later, the patient developed new skin lesions and worsening bilateral leg edema. There were multiple small erythematous and hemorrhagic papules, macules, and blisters on the medial aspect of the right lower leg and ankle, each measuring less than 1 cm in diameter (Figure 1). The clinical differential diagnosis included vasculitis related to an underlying collagen vascular disease, atypical edema blisters, and drug hypersensitivity reaction. A punch biopsy of one of the lesions showed a moderately dense superficial and deep perivascular lymphoid infiltrate with marked papillary dermal edema and early subepidermal split (Figure 2). The infiltrate was comprised of small- to medium-sized lymphocytes admixed with large cells, histiocytes, and plasma cells (Figure 3). Immunohistochemistry revealed a predominance of CD3+ and CD4+ small- to medium-sized T cells. CD20 highlighted the large angiocentric B cells (Figure 4), which also were positive on EBV-encoded small RNA (EBER) in situ hybridization (Figure 5). A diagnosis of LYG was rendered. Approximately 40 to 50 EBV-positive large B cells were present per high-power field (HPF), consistent with grade 2 disease.
,
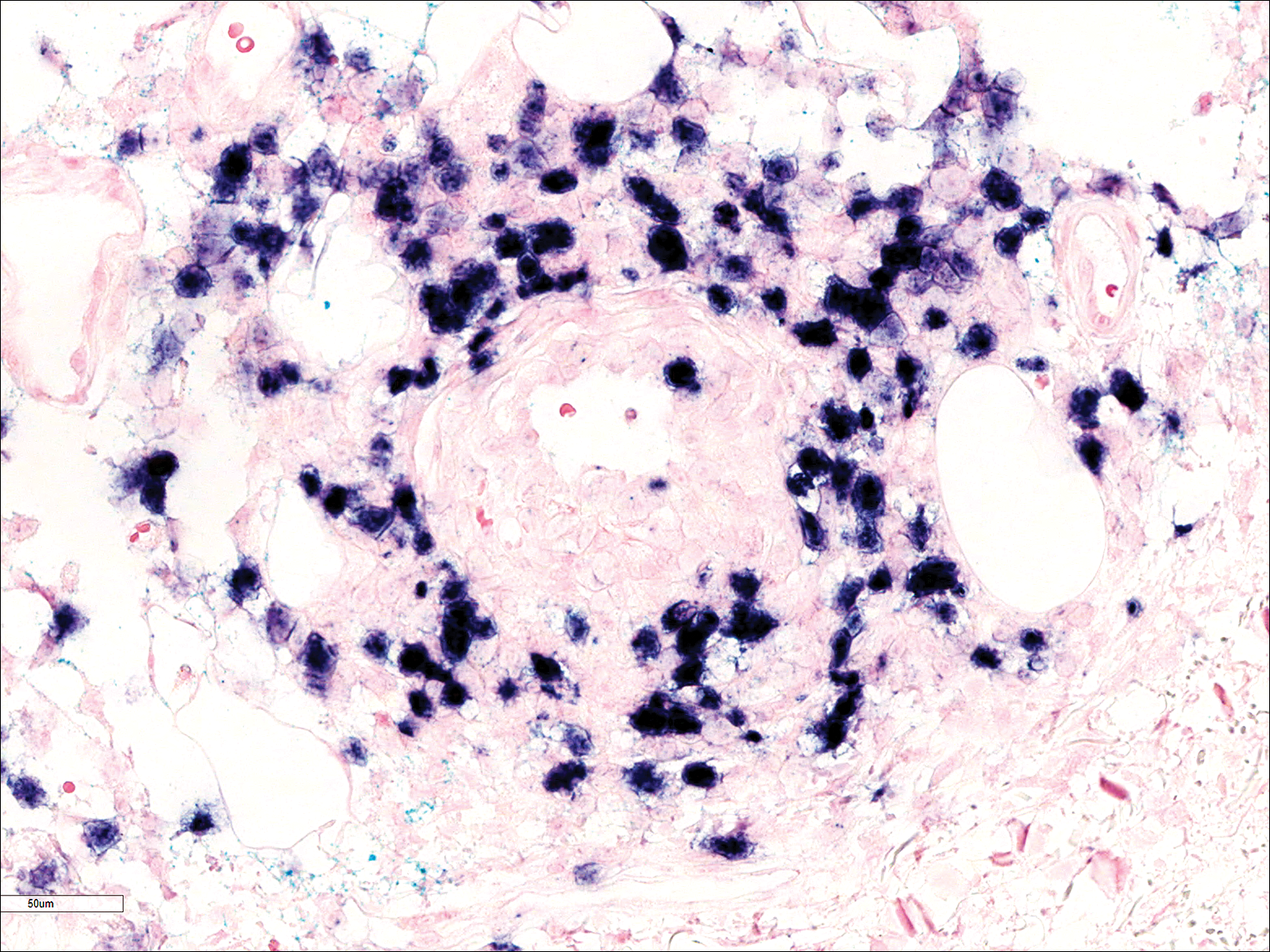
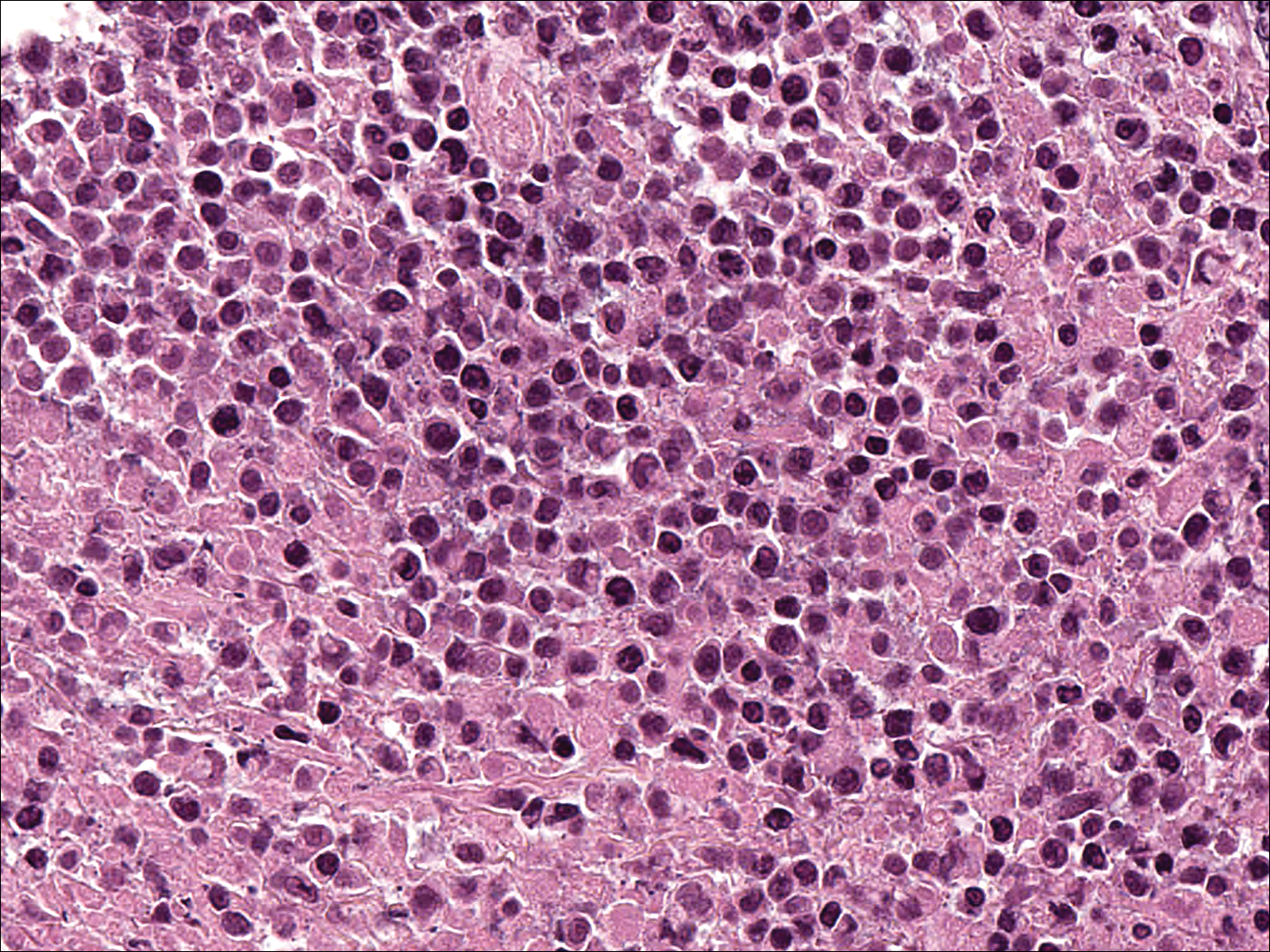
Soon after diagnosis, follow-up computed tomography of the chest, abdomen, and pelvis revealed suspicious lesions in the kidneys, liver, spleen, and inguinal and iliac lymph nodes. The ground-glass opacities in the lungs continued to progress, with 2 additional nodules noted in the right upper and lower lobes. Four days later, core needle biopsies of the right inguinal lymph node showed a large B-cell lymphoma with extensive necrosis (Figure 6). EBER in situ hybridization was suboptimal, probably due to extensive necrosis.
She was started on etoposide, prednisolone, vincristine, cyclophosphamide, and doxorubicin (EPOCH) for 5 days before developing Klebsiella pneumoniae sepsis and acute kidney injury. She was transferred to the critical care unit due to increasing oxygen requirement. Despite medical interventions, she continued to decompensate and elected to transition to palliative care. She died 6 weeks after the initial presentation. Her family did not request an autopsy.