Lichen Sclerosus of the Eyelid





Practice Points
- Lichen sclerosus is not confined to only the anogenital area and can affect the face in rare cases.
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
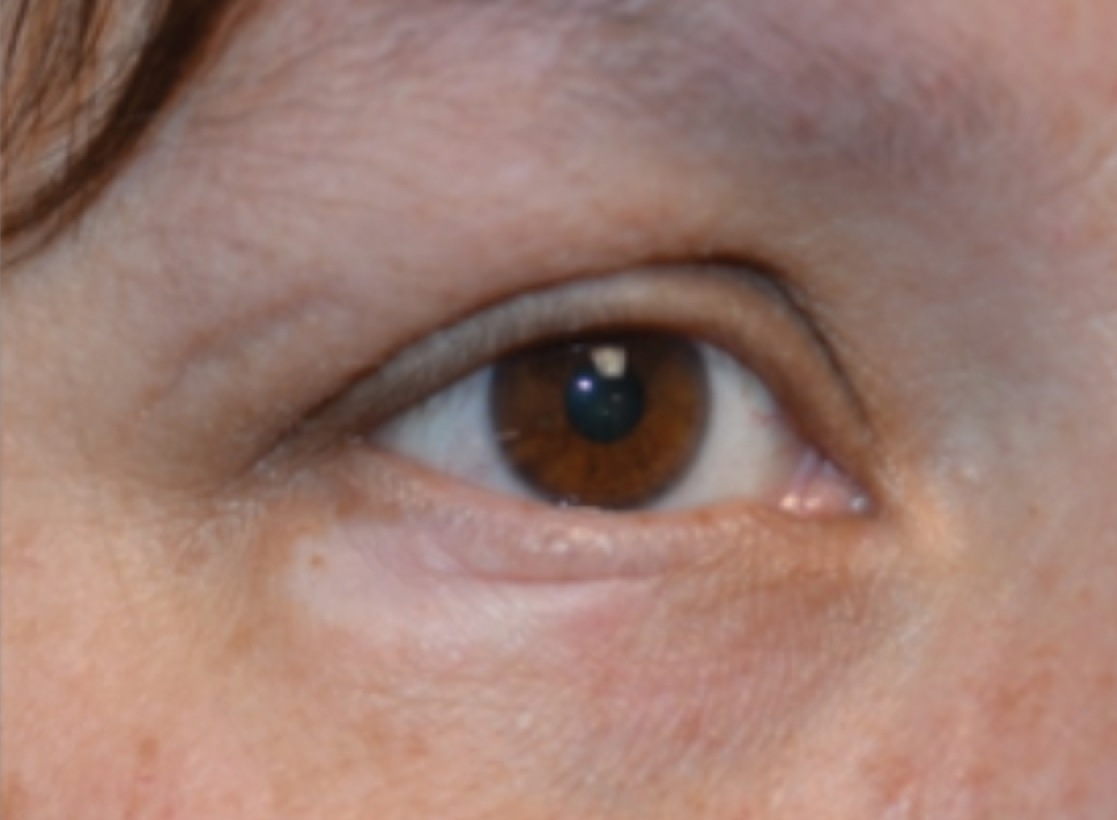
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
,
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
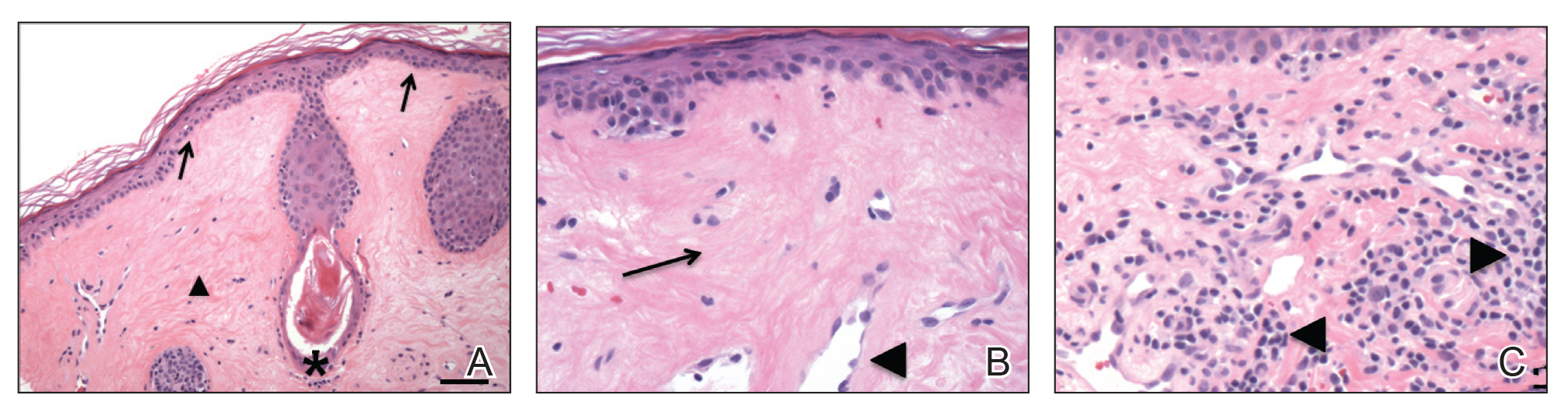
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.