Annular Elastolytic Giant Cell Granuloma: Mysterious Enlarging Scarring Lesions





Practice Points
- Annular elastolytic giant cell granuloma (AEGCG) should be kept in the differential diagnosis when assessing a middle-aged woman with recurring annular plaques with a raised border and an atrophic center on both sun-exposed and sun-protected areas of the body.
- Histologically, AEGCG classically has a granulomatous component in the dermis that lacks elastic tissue and has no necrobiosis, lipids, or mucin. Staining with elastin may be necessary to highlight these areas as well as demonstrate elastophagocytosis.
To the Editor:
A 52-year-old woman with a medical history of migraines and cervicalgia presented with lesions on the right arm, back, and right calf. The patient stated that the lesions began as small papules that had grown over 13 months, with the largest papule on the right forearm. She reported no itching, bleeding, pain, discharge, or other symptoms associated with the lesions. She had a multiple-year history of similar lesions that did not respond to treatment with antifungals, moderate-potency steroids, and other over-the-counter creams. The lesions would resolve spontaneously with scarring and subsequently recur. Prior skin biopsies were inconclusive. The patient did not report any systemic symptoms or a personal or family history of connective tissue diseases.
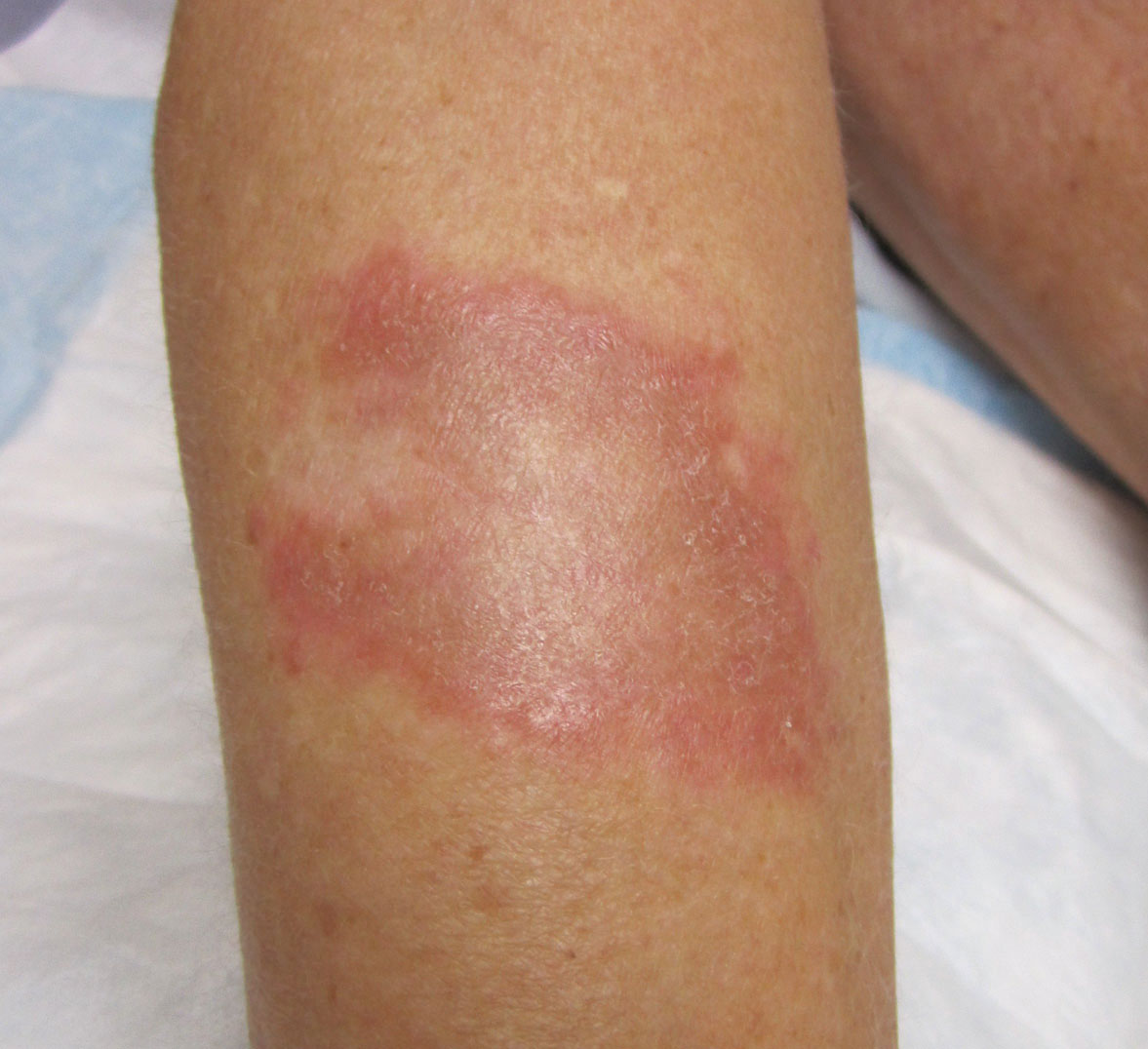
Physical examination revealed a 4-cm asymmetric, annular, erythematous plaque with central clearing on the right dorsal forearm with defined margins except over the distal aspect (Figure 1). She also had several 1- to 2-cm erythematous, nummular, asymmetric plaques on the right upper arm with well-defined margins. She had several lesions over the central and left sides of the upper back that were similar to the lesions on the upper arm.
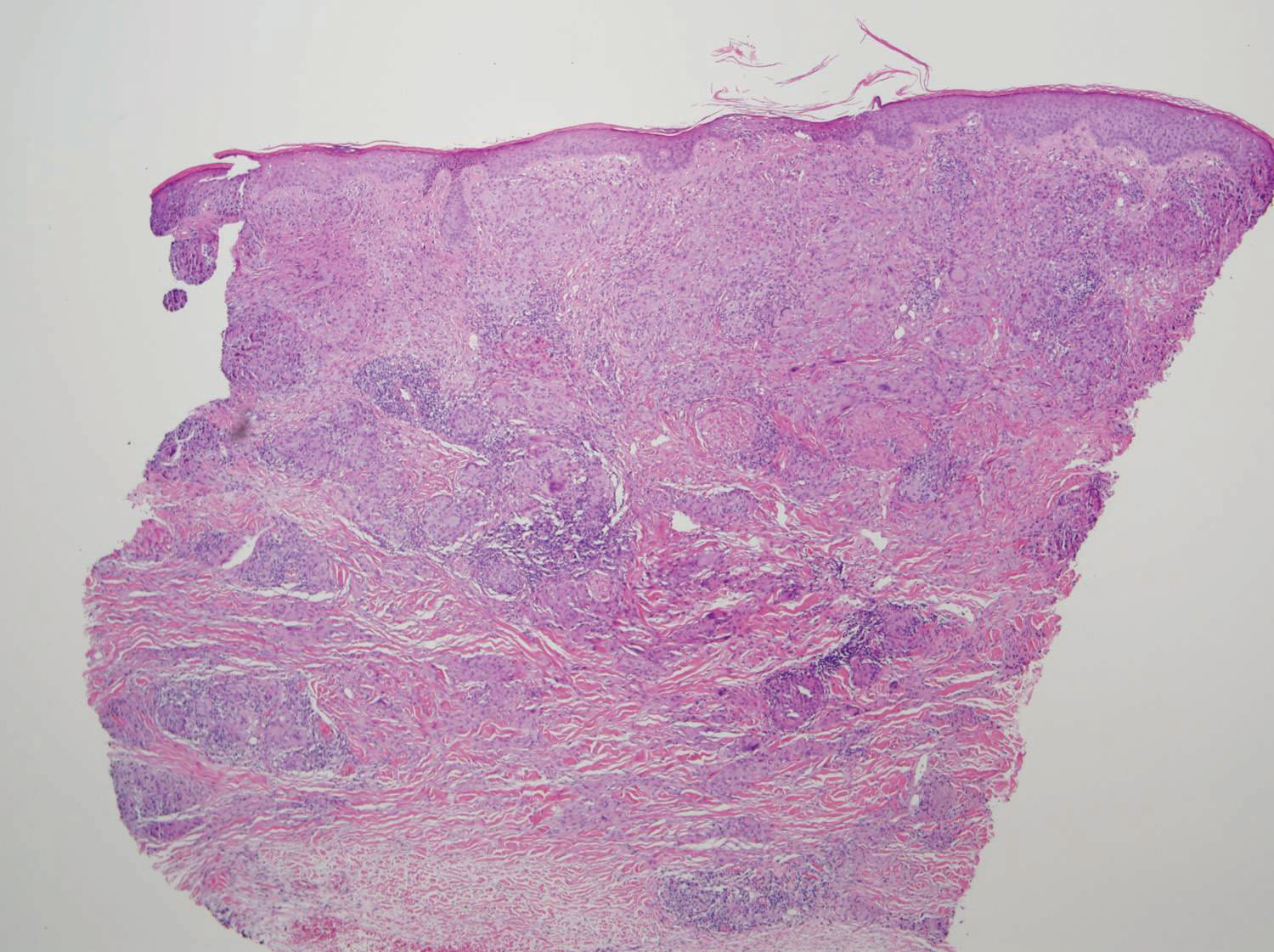
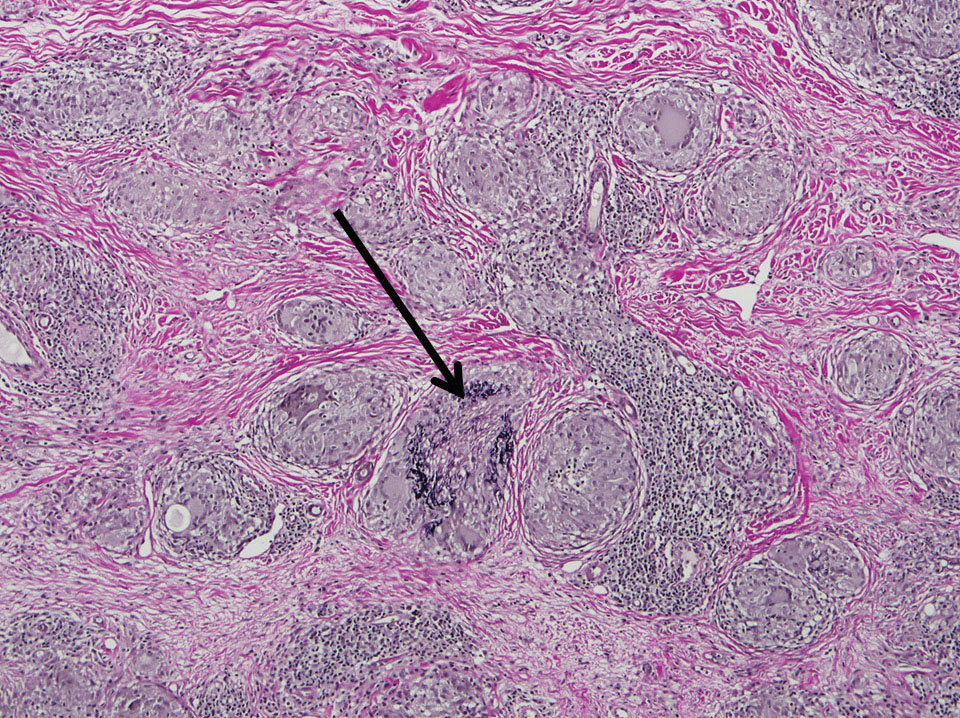
Two 4-mm punch biopsies of the right dorsal forearm and left side of the upper back revealed similar histologic features with a predominantly unremarkable epidermis. The dermis revealed a lymphohistiocytic infiltrate with prominent multinucleated giant cells organized into foreign body–type granulomas that extended into the deep dermis and subcutaneous tissue (Figure 2). In the granulomatous areas, there was a near-complete loss of elastic fibers with focal elastophagocytosis highlighted with Verhoeff-van Gieson (elastin) stain (Figure 3). Grocott-Gomori methenamine-silver and Fite stains for microorganisms were negative, and there was an absence of necrobiosis, lipids, and mucin.
,
The histologic findings of a granulomatous dermatitis with loss of elastic fibers and elastophagocytosis in addition to the patient’s clinical presentation and history were consistent with the diagnosis of annular elastolytic giant cell granuloma (AEGCG). Infectious and other granulomatous diseases including sarcoidosis were ruled out via clinical history, unremarkable laboratory analysis (ie, complete blood cell count, chemistry panel, antinuclear antibody, urinalysis), and a normal chest radiograph. The histologic findings via the various stains were instrumental to the diagnosis. The patient was treated with fluocinonide and subsequently lost to follow-up.
Annular elastolytic giant cell granuloma is an uncommon cutaneous disease that presents with recurring annular plaques with raised erythematous borders and subsequent residual scarring.1 O’Brien2 originally described this condition in 1975 as an actinic granuloma due to similar histologic findings in areas of the patient’s sun-exposed skin. Ragaz and Ackerman3 disputed O’Brien’s2 description, claiming granulomatous inflammation was a primary pathologic process and not a consequence to damaged elastotic material. In 1979, Hanke et al4 termed the lesions as AEGCG because he did not find a correlation to the sun-exposed areas of the patients and did not see solar elastosis.
Although AEGCG has an unclear pathogenesis, cellular immunologic reactions induced by modified function of elastic fibers’ antigenicity contribute to AEGCG formation.5 Therefore, environmental and host factors may play a role in its etiopathogenesis. In one study, 37% of 38 Japanese patients with AEGCG were found to have definitive or latent diabetes mellitus, raising the possible role of diabetes in the structural damage of the elastic fibers.6
Patients typically are middle-aged women who present clinically with red or atrophic plaques that have slightly elevated borders. They have centripetal spread with a resulting atrophic center.7 Clinically, the differential diagnosis of this condition includes actinic granuloma, granuloma annulare, and granuloma multiforme.8
Histologically, AEGCG has a granulomatous component with multinucleated giant cells in the upper and mid dermis. This component typically is distributed peripherally to a central zone that lacks elastic tissue. Elastophagocytosis, a classic finding in AEGCG, is the phagocytosis of elastic fibers that can microscopically be seen in the cytoplasm of histiocytes and multinucleated giant cells. There also is an absence of necrobiosis, lipids, mucin, and a palisading arrangement of the granulomas. These findings distinguish AEGCG from granuloma annulare and necrobiosis lipoidica, the primary histologic differential diagnoses.9 In addition, consideration of entities consistently exhibiting elastophagocytosis such as mid-dermal elastolysis, papillary dermal elastolysis, actinic granuloma, and granulomatous slack skin should be considered.5,10,11
Therapy for AEGCG is broad and includes topical, intralesional, and systemic corticosteroids. Hydroxychloroquine, isotretinoin, clofazimine, dapsone, photochemotherapy, and cyclosporine also have been utilized with varying results. Other reports show improvement with surgical excision, cryotherapy, or cauterization of small lesions.12-15