Do glutamatergic drugs have a role in treating depression?






Mrs. S, age 46, has been struggling to manage depression for 7 years. She completed adequate trials of several selective serotonin reuptake inhibitors and bupropion. Currently, she is taking duloxetine, 60 mg/d, and aripiprazole, 5 mg/d.
At her most recent clinic visit, Mrs. S reports that she is doing “OK,” but that she still feels sad and disengaged most days of the week. She wants to know more about ketamine for treating depression after reading about it on the Internet and hearing it mentioned in a support group she attends. She asks if you think it would work for her, and gives you with a copy of an article about its use in patients with treatment-resistant depression. Mrs. S has no other health conditions and takes a daily vitamin D and calcium supplement.
The monoamine hypothesis of depression postulates that symptoms originate from underactivity of monoamines, such as serotonin, norepinephrine, and dopamine, in the brain. This hypothesis was formulated in the 1960s after researchers observed that monoamine oxidase inhibitors and tricyclic antidepressants relieved depressive symptoms; both were known to increase monoamine concentrations in the synaptic cleft.1

Regrettably, these medications do not adequately relieve depressive symptoms for many people. In fact, symptom remission occurs in only one-third of treated patients.2 This low remission rate reflects a lack of understanding of the pathophysiology of depression, and the need for drugs with unique mechanisms of action.
One of the newest drug targets shown to be relevant in psychiatric illness is the
glutamatergic system. Glutamate is the predominant excitatory neurotransmitter in the CNS, and it is responsible for many key functions, including synaptic plasticity, learning, memory, and locomotion.3 Normally, the glutamatergic system tightly regulates the amount of glutamate in the neuronal synapse via receptors on presynaptic and postsynaptic neurons, as well as on glial cells (Figure). When this equilibrium is disrupted in stressful situations, such as ischemia, trauma, or seizures, excess glutamate is released into the synapse. The resulting glutamatergic hyperactivity can lead to neurotoxicity and cell death when neuronal receptors are activated for an extended period. 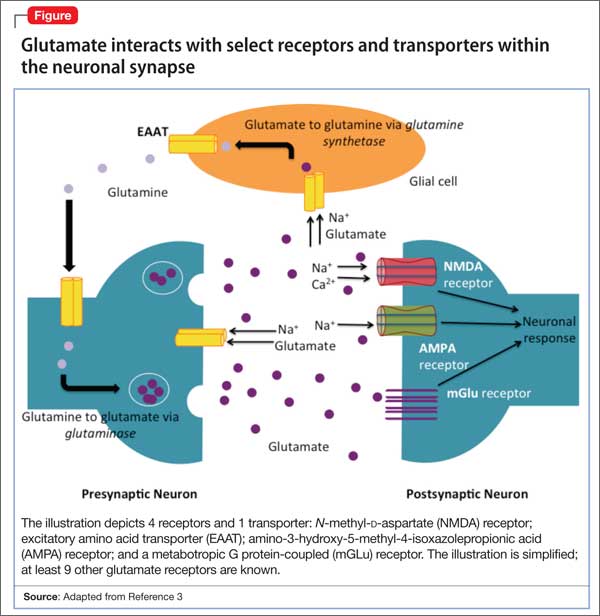
A key component of the glutamatergic system that is responsible for removing excess glutamate from the synapse is membrane-bound transporters, which are similar to serotonin and norepinephrine transporters. These excitatory amino acid transporters (EAATs) are important because glutamate metabolism does not occur within the synapse and EAATS are responsible for removing most of the glutamate from the synapse into glial cells.3
The network of receptors within the synapse that are activated by glutamate is extensive and complex. There are at least 11 glutamate-responsive receptors: 3 are ionotropic action channels, and the remaining 8 are metabotropic G protein-coupled receptors. Previous studies have shown regional changes in glutamate receptors, as well as elevated levels of glutamate, in the brains of patients with major depressive disorder (MDD).4
Ketamine. The ionotropic receptor N-methyl-d-aspartate (NMDA) is one of the most studied glutamate receptors. Pharmacologically, ketamine is a noncompetitive NMDA receptor antagonist that also activates the amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor, which is another subtype of ionotropic glutamate receptors. In open-label clinical trials, ketamine has demonstrated rapid antidepressant action in patients with treatment-resistant MDD.4,5
Recently, Murrough et al6 performed the first randomized, psychoactive controlled trial using a single IV infusion of ketamine dosed below anesthesia ranges (0.5 mg/kg), or midazolam (0.045 mg/kg), in patients with treatment-resistant depression who had been antidepressant-free for at least 4 weeks. They found that 24 hours after medication administration, the likelihood of response to ketamine was significantly higher than the response to midazolam (OR: 2.18; 95% CI: 1.21 to 4.14), with a response rate of 64% in the ketamine group and 28% in the midazolam group.6
Psychotropic side effects, such as hallucinations, are a major concern with ketamine tolerability and abuse potential. This is largely because of ketamine’s antagonism of the NMDA receptor, which is a property shared with other abused drugs such as phencyclidine (PCP) and dextromethorphan. In the Murrough et al6 study, there were no reported cases of paranoia or hallucinations, but dissociative symptoms were relatively common (17%).
Although the results in this trial appear encouraging, there are several limitations to using ketamine to treat MDD, especially in an ambulatory setting. Concerns include ketamine’s IV administration, potential for abuse, long-term efficacy, and side-effect profile—particularly psychotic symptoms and hemodynamic changes. An ideal compound would have the rapid efficacy of ketamine, but with a safer side-effect profile, easier administration, and less potential for abuse.
Riluzole also acts on the glutamatergic system, but has not shown antidepressant efficacy as consistently as ketamine. Riluzole is FDA-approved for treating amyotrophic lateral sclerosis.5 Pharmacologically, riluzole is a glutamatergic modulator that increases glutamate reuptake into glial cells, decreases glutamate release, and increases AMPA trafficking. In open-label studies riluzole has shown efficacy in reducing depressive symptoms.4,5 However, when compared with placebo as a means of sustaining treatment response after a 1-time dose of ketamine, riluzole showed was no significant improvement in time to depressive relapse.7