Glutamate: New hope for schizophrenia treatment






Research on glutamatergic dyfunction may lead to therapies targeting negative and cognitive symptoms
Discuss this article at www.facebook.com/CurrentPsychiatry
In patients with schizophrenia, positive symptoms typically respond to treatment, while negative and cognitive symptoms often persist and contribute to chronic disability.1 Schizophrenia also is associated with widespread neurocognitive deficits—including impairments in executive functioning, learning, memory, and processing speed—that are a core feature of the disorder and may precede illness onset.2
Current treatment is based on the dopamine model of schizophrenia, which proposes that dopaminergic dysfunction is the basis for symptoms and cognitive deficits.3 Although this model is effective in guiding treatment for some patients, most show persistent disability despite receiving the best available treatment. Over the last 2 decades, researchers have developed alternative conceptual models of schizophrenia based on the psychotomimetic effects of compounds such as phencyclidine (PCP) and ketamine.4 These compounds function primarily by blocking N-methyl-D-aspartate (NMDA)-type glutamate receptors (NMDARs), which has lead researchers to focus on glutamatergic neurotransmission and NMDARs as a basis for new drug development. This article describes the glutamatergic model of schizophrenia and its implications for future treatments.
Dopaminergic models
Since the discovery of chlorpromazine almost 60 years ago, the dopamine model of schizophrenia has been widely accepted. It has gone through several iterations but in general suggests that schizophrenia is caused by dopaminergic system dysfunction, particularly increased dopamine within subcortical brain regions such as the striatum or nucleus accumben.3 The ability of amphetamine or other dopaminergic agents to induce symptoms closely resembling positive symptoms supports this model, as do genetic studies that show dopamine-related genes are associated with schizophrenia.5 In addition, all antipsychotics block dopamine type 2 receptors.
Unfortunately, limitations of this model continue to limit treatment:
- Dopaminergic compounds such as amphetamine do not induce negative symptoms or cognitive deficits similar to those observed in schizophrenia.
- Dopamine receptor blockers do not reverse cognitive dysfunction or negative symptoms.
- Dopaminergic instability observed during acute decompensation appears to resolve after stabilization even without symptom remission.
- Although dopaminergic systems preferentially innervate frontal brain regions, cognitive deficits in schizophrenia appear to be widespread, involving sensory as well as frontal brain systems.
Thus, dopaminergic dysfunction appears to account for only a part of schizophrenia’s symptomatic and neurocognitive profile.
Glutamatergic model
Approximately 20 years ago, researchers proposed an alternate schizophrenia model based on the observed clinical actions of “dissociative anesthetics,” including PCP and ketamine. PCP was patented in 1953 as a surgical anesthetic, but serious side effects, such as hallucinations, agitation, and catatonic-like reactions, soon curtailed its clinical use. As early as 1959, some researchers noted similarities between PCP psychosis and schizophrenia.4,6
The binding site for PCP and other dissociative anesthetics (“PCP receptor”) was first described in 1979 and subsequently localized within the ion channel formed by the NMDAR. Glutamate is the primary excitatory neurotransmitter in the brain, and binds to NMDA and non-NMDA (eg, metabotropic or alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid [AMPA]) receptors. Binding of PCP prevents glutamate from activating NMDARs, which suggests that the pathogenesis of schizophrenia may be caused by dysfunction of NMDARs in particular or of the glutamatergic system in general. Unlike dopamine, the glutamatergic system is distributed throughout the brain and plays a prominent role in sensory processing and higher-level functions such as memory and executive functioning (Figure).6 Therefore, glutamatergic theories open new approaches for potential schizophrenia treatments, most of which are now entering clinical evaluation.
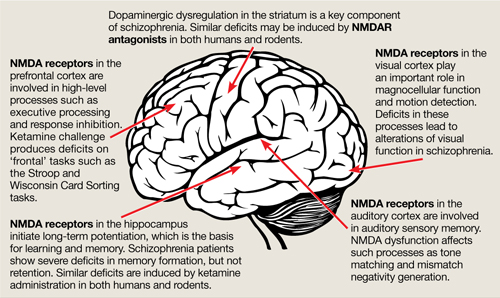
Figure: The wide reach of glutamatergic dysfunction
NMDA: N-methyl-D-aspartate; NMDAR: N-methyl-D-aspartate-type glutamate receptors
Source: Reference 6
Effects of NMDAR antagonists
In initial studies with PCP and ketamine in the early 1960s, researchers noted that these agents produced psychotic effects similar to schizophrenia symptoms.6 Further confirmation was obtained from retrospective studies of PCP abusers.6 It was not until the 1990s, however, that studies using modern operationalized symptom and neuropsychological rating scales were conducted. In those studies, healthy participants developed positive symptoms, negative symptoms, and cognitive dysfunction after receiving ketamine.7,8 Moreover, in these studies the balance between negative and positive symptoms was similar to that typically observed in schizophrenia, as was the pattern of cognitive dysfunction. Therefore, unlike dopaminergic agents, NMDAR antagonists appear to be able to produce the full constellation of symptoms and cognitive deficits associated with schizophrenia.
Similarly, ketamine worsened positive and negative symptoms in patients diagnosed with schizophrenia.9 Although acute challenge with NMDAR antagonists does not produce schizophrenia-like auditory hallucinations in healthy controls, it does induce sensory distortions similar to those seen in individuals with early schizophrenia and does exacerbate pre-existing hallucinations in schizophrenia patients.10 Thus, acute challenge with NMDAR antagonists appears to re-create a state similar to the earliest stages of schizophrenia.6