
How to diagnose and repair congenital uterovaginal malformations






Knowing when and how these abnormalities become apparent is the first step toward correcting them
IN THIS ARTICLE
Repair involves incision of the membrane near the hymeneal ring, followed by evacuation of accumulated blood. The cervix may appear to be flush with the vaginal apex, owing to compression, but this condition usually corrects itself within 2 or 3 weeks. Extra hymeneal tissue is excised to create an orifice of normal size, and the vaginal mucosa is intermittently sutured to the hymeneal ring using absorbable material (FIGURE 3).
FIGURE 2 Imperforate hymen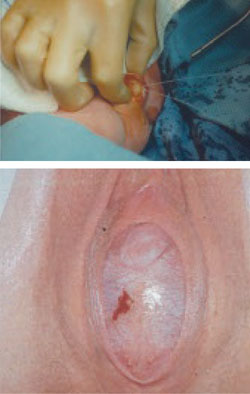
Bulging hymen due to mucocolpos in a newborn (top) and hematocolpos in an adolescent (bottom).
FIGURE 3 Hymenectomy restores vaginal orifice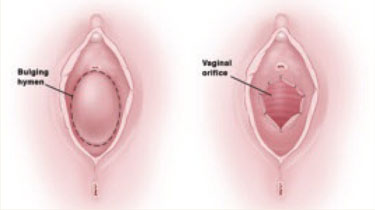
Extra hymeneal tissue is excised to create an orifice of normal size. The vaginal mucosa is intermittently sutured to the hymeneal ring.
Incomplete hymeneal fenestration
This condition often goes undiagnosed until the patient seeks evaluation for an inability to insert tampons or medication or because of coital difficulty. Young girls with microperforate hymen may present with postmenstrual spotting or malodorous discharge due to partial obstruction and poor drainage.
Treatment of microperforate, cribriform, or septate hymen entails resection of excess hymeneal tissue to create a functional hymeneal ring.
Transverse vaginal septum
A transverse septum represents failed canalization of the vaginal plate. This condition occurs in approximately one of every 18,000 to 30,000 women.1,2 The septum can be located at any of various levels in the vagina; approximately 46% are found in the upper vagina, 35% to 40% in the middle portion, and 15% to 20% in the lower vagina.1,2 Lower septa appear to be thinner than those in the proximal vagina; thinness may be due to the pressure of accumulated fluid. If a small perforation is present, the patient will experience irregular spotting.
Examination. The external genitalia appear to be normal, but the vagina is shortened. Children may present with mucocolpos, whereas adolescents develop hematocolpos. If a small perforation is present, pyohematocolpos may result from ascending infection. A mass can be palpated on rectal examination.
Ultrasonography (US) or magnetic resonance imaging (MRI) helps define the location and thickness of the septum and distinguish a high septum from congenital absence of the cervix.
Treatment. Resect a small septum and perform end-to-end anastomosis of the upper and lower edges of the vaginal wall (FIGURE 4). Only an experienced surgeon should excise a thick septum. Reanastomosis is easier if the upper vagina is distended with menstrual blood. To ensure healing without stricture, mobilize the vaginal edge circumferentially before suturing the raw edges.
Outflow obstruction caused by a transverse septum in the distal vagina almost never leads to hematometra or hematosalpinx. However, septa that arise in the proximal vagina are usually thicker and, if left untreated, can cause reflux that damages the upper reproductive tract. These cases are almost always associated with severe dysmenorrhea and should be referred to an experienced vaginal surgeon. Pelvic MRI is recommended to assess the thickness and precise anatomy of the defect before repair.
FIGURE 4 Removing a transverse vaginal septum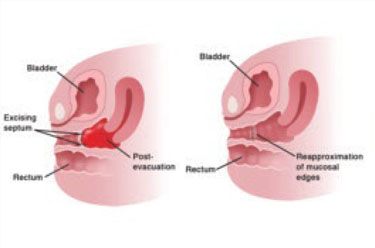
The septum is resected from the distal vagina. This is followed by end-to-end anastomosis of mucosal edges.
Uterine septum
A septate uterus has a normal external surface but two endometrial cavities. It is caused by a defect in fusion or resorption of the midline septum between the two müllerian ducts. The degree of septation varies from a small midline septum to a total failure of resorption that produces a septate uterus with a longitudinal vaginal septum. A slight midline septum with minimal fundal indentation is classified by some as a septate uterus; by others, as a bicornuate uterus.
A higher risk of recurrent miscarriage is associated with longer septa.
Surgical repair with hysteroscopic resection of the septum yields excellent results.
CASE 2 The problem of agenesis
An 18-year-old patient consults a gynecologist because she has never menstruated and is unable to have sexual intercourse. She appears to be phenotypically normal, with normal labia upon examination, but no hymeneal opening is found. Pelvic US reveals absence of the uterus with ovaries that appear to be normal.
How should her condition be managed?
Uterovaginal agenesis, also known as müllerian aplasia or Mayer–Rokitansky–Küster–Hauser (MRKH) syndrome, refers to congenital absence of the vagina with variable uterine development (FIGURE 5). It arises from agenesis of the
müllerian duct system, although the underlying cause is unknown. Vaginal agenesis is usually accompanied by cervical and uterine agenesis. However, 7% to 12% of patients with this syndrome have a normal but obstructed or rudimentary uterus with functional endometrium. This condition occurs in roughly one of every 5,000 females.1,3
In addition, these girls often exhibit extragenital anomalies.1,4 Approximately 25% to 50% have a urologic anomaly, such as unilateral renal agenesis, pelvic or horseshoe kidneys, or irregularities of the collecting systems; 10% to 15% have another type of anomaly, such as congenital heart disease, abnormalities of the hands, congenital deafness, cleft palate, and inguinal or femoral hernia.
