Novel gene-based therapies for neuromuscular diseases






Neuromuscular diseases (NMDs) are a broad classification of heterogeneous groups of disorders characterized by progressive muscle weakness resulting from muscle or nerve dysfunction.1 Diagnosis is based on symptoms and a full medical history, as well as on muscle and imaging tests (including electromyography, nerve-conduction studies, magnetic resonance imaging, muscle biopsy, and blood tests) to confirm or rule out specific NMDs.2 Early diagnosis of NMDs can be difficult because symptoms overlap with those of many other diseases.
Although individually, NMDs are rare, collectively, they affect approximately 250,000 people in the United States. Disease types vary in regard to cause, symptoms, prevalence, age of onset, progression, and severity. Functional impairment from any NMD can lead to lifelong morbidities and shortened life expectancy.1,3
Treatment options for NMDs are limited; most target symptoms, not disease progression. Although there is a need for safe and effective gene-based therapies for NMDs, there are challenges to developing and delivering such treatments that have impeded clinical success. These include a lack of understanding about disease pathology and drug targets, limited animal model systems, and few reliable biomarkers that are predictive of therapeutic success.4,5

Notwithstanding that challenges remain, our understanding of gene expression in NMDs has greatly advanced in the past few decades. This progress has translated into promising results in the gene-therapy field – thereby setting the stage for therapeutic approaches that use novel gene-delivery and gene-manipulation tools.6 These novel approaches include nonviral strategies, such as antisense oligonucleotides (ASOs), and viral-based strategies, such as adeno-associated virus (AAV)-mediated gene silencing and AAV-mediated gene delivery.
In this article, we highlight advancements in the clinical development of gene-based therapies for NMDs. We focus on amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), and Duchenne muscular dystrophy (DMD) because of recent clinical successes in developing such therapies.1,6,7 We also catalog completed and ongoing clinical trials for ALS, SMA, and DMD (Tables 1-3).
Amyotrophic lateral sclerosis
ALS is caused by progressive degeneration of upper- and lower-motor neurons, which eventually leads to respiratory failure and death 3 to 5 years after disease onset.7-9 There are two subtypes: Familial ALS (10% of cases) and sporadic ALS (90% of cases). Commonly mutated ALS-associated genes6,8 are:
- Superoxide dismutase type 1 (SOD1).
- Chromosome 9 open reading frame 72 (C9orf72).
- Transactive response DNA-binding protein 43 (TARDBP).
- Fused in sarcoma (FUS).
SOD1-targeted therapy is being studied, with early evidence of clinical success. Mutations in SOD1 account for 10% to 20% of familial ALS cases and 1% to 2% of sporadic ALS cases.6,10 10 Mutations in C9orf72 account for 25 to 40% of familial ALS cases and 7% of sporadic ALS cases.8,9,11 Mutations in TARDBP account for 3% of familial ALS cases and 2% of sporadic cases.12 Mutations in FUS account for 4% of familial ALS cases and 1% of sporadic cases. Overall, these mutant proteins can trigger neurotoxicity, thus inducing motor-neuron death.6,10
Treatment of ALS
Two treatments for ALS are Food and Drug Administration approved: riluzole (Rilutek), approved in 1995, and edaravone (Radicava), approved in 2017.

Riluzole is an oral anti-excitotoxic glutamate antagonist.11 Approval of riluzole was based on the results of two studies that demonstrated a 2- to 3-month survival benefit.10,14 For patients who have difficulty swallowing, an oral suspension (Tiglutik, approved in 2018) and an oral film (Exservan, approved in 2019) are available.
Edaravone is a free-radical scavenger that decreases oxidative stress and is administered intravenously (IV).9,13,14 Findings from clinical trials suggest functional improvement or slower decline in function for some patients.
Although these two agents demonstrate modest therapeutic benefit, neither reverses progression of disease.10,14
Gene-based therapy for ALS
Many non-viral strategies, including antisense oligonucleotide (ASO), monoclonal antibodies, reverse transcriptase inhibitors, and HGF gene replacement therapy are used as therapeutic approaches to SOD1, C9orf72, and FUS gene mutations in ALS patients, and are being evaluated in clinical studies14,15 (Table 113-17).
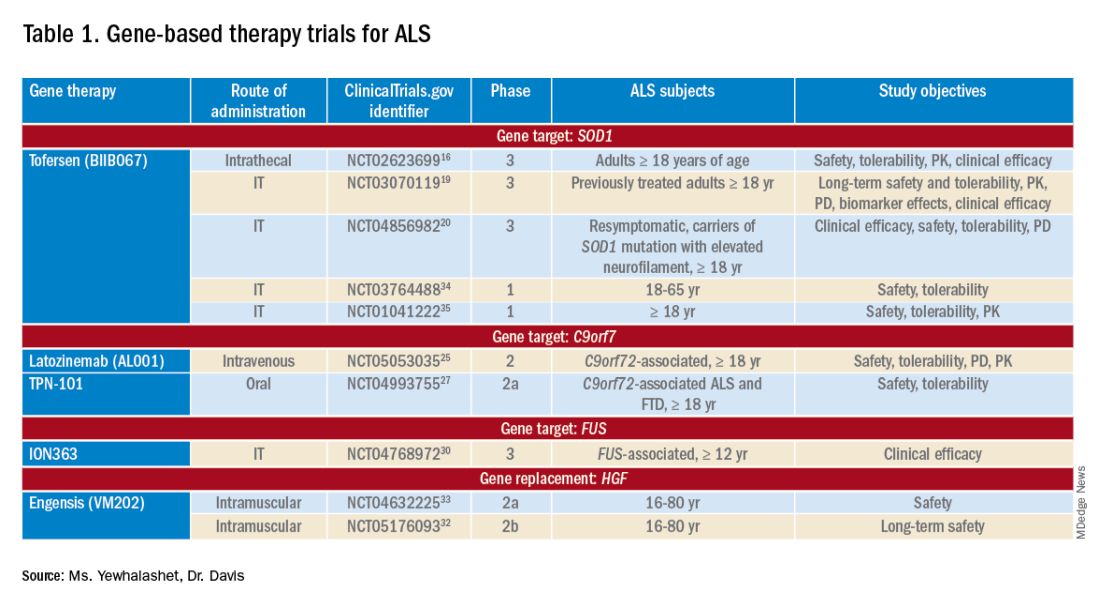
Tofersen, also known as BIIB067, is an investigational ASO, administered by intrathecal (IT) injection, that binds to SOD1 mRNA, thus reducing its protein levels.16 Tofersen was evaluated in the VALOR phase 3 study (ClinicalTrials.gov Identifier: NCT02623699), a three-part randomized, double-blind, placebo-controlled trial: single ascending dose (Part A), multiple ascending dose (B), and fixed dose (C).10 In Parts A and B, 48 participants received five IT injections of tofersen or placebo over 12 weeks and were followed for an additional 12 weeks. Reduction in SOD1 protein production and neurofilament level in cerebrospinal fluid (CSF) (a potential biomarker of motor-neuron degeneration) was observed, which determined the fixed-dose for Part C.16,17
Part C examined the efficacy, safety and tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of tofersen, compared with placebo, in adults with ALS who had a confirmed SOD1 mutation.17 A total of 108 participants were enrolled; 60 were identified as “faster-progressing”; 48, as “slower-progressing.”18 The primary endpoint of Part C was change from baseline to Week 28 on the Revised ALS Functional Rating Scale (ALSFRS-R) total score. (ALSFRS-R measures overall clinical effect; the score ranges from 0 [no function] to 4 [full function].17)
Tofersen failed to meet the primary efficacy outcome because statistically significant findings were lacking in the faster-progressing population, as measured by joint-rank analysis (difference of 1.2 on the ALSFRS-R score; P = .97). However, trends favoring tofersen were observed across key secondary clinical outcome measures18:
- Change from baseline in CSF SOD1 protein concentration.17 Percent reduction in the total SOD1 protein level was much higher in the tofersen-treated group than in the control group (38% more than controls in the faster-progressing population; 26% more than controls in the slower-progressing population).18
- Change from baseline in neurofilament light-chain concentration in plasma.17,18 Percent reduction in the level of neurofilament light chain was also observed to be higher in the tofersen-treated group than in the control group (67% more than controls in the faster-progressing population and 48% more than controls in the slower-progressing population).18