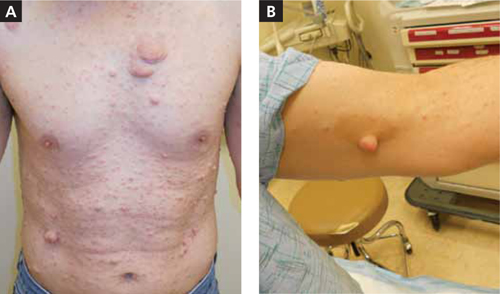
Painless cutaneous nodules






The lesions on the patient’s body were getting caught on his clothes. He said that his mother and several of his siblings had similar “lumps.”
Diagnosis: Neurofibromatosis type 1
Neurofibromatosis type 1 (NF1) is an autosomal dominant multisystem disorder affecting about one in 3000 people.1-3 The NF1 gene is on chromosome 17q11.2, which encodes for neurofibromin protein.2,3 About half of these patients demonstrate a spontaneous mutation.1-5 Clinical features include cutaneous, subcutaneous, skeletal, peripheral, and central nervous systems abnormalities.
3 common features
Neurofibroma is the hallmark of NF1. Cutaneous and subcutaneous neurofibromas are benign peripheral nerve sheath tumors consisting of Schwann cells, fibroblasts, peripheral cells, mast cells, axons, and blood vessels.1-5 Neurofibromas usually develop when patients are in their late teens; they are often painless.
Café-au-lait spots are well-circumscribed, evenly pigmented, light to dark brown macules and/or patches with an average size of 2 to 5 cm (FIGURE 1B). Up to 10% of the general population may have one or 2 café-au-lait spots with no other abnormalities.6 Patients with NF1 will have 6 or more.1
Skin fold freckling is the most specific feature in patients with NF1. This freckling usually occurs in the axilla and groin regions when patients are between 3 and 5 years of age. The freckles are typically small, with an average size of 1 to 3 mm.
Other less common clinical features of NF1 include plexiform neurofibroma, skeletal abnormalities (short stature, scoliosis, long bone dysplasia, and osteopenia/ osteoporosis), Lisch nodules (iris hamartomas), neurocognitive deficits, cardiovascular abnormalities, and optic pathway gliomas.1-5
FIGURE 1
Widespread cutaneous nodules with hyperpigmented patches