Genetic blood disorders: Questions you need to ask






No one is better positioned than you to look for evidence of inherited diseases—especially when you consider that many FPs care for 2, or even 3, generations of a single family.
Clinical presentation
Mild forms of thalassemia are often mistaken for iron deficiency anemia, and it is likely that the prevalence of this inherited disease is underestimated.19 Patients at the intermediate level often need transfusions under circumstances in which they’re not typically required, such as childbirth. Those with thalassemia major develop splenomegaly and bone malformations. Although patients with thalassemia major have not been expected to live much beyond adulthood, the disorder has been treated successfully with bone marrow transplantation in recent years.20
Treatment and follow-up
Patients who are severely affected by thalassemia need frequent transfusions; ensuring that they receive emergency services, as needed, is key. When caring for such patients—or for individuals who have symptoms suggestive of thalassemia or whose children are found to be carriers—targeted questions about family medical history are necessary, as well. Asking whether anyone in the family has required blood transfusions or had “problems with their blood” may help you detect patterns suggestive of a family history of thalassemia.
As is the case with sickle cell trait, however, the significance (or evidence) of carrier status may be lost by the time a child reaches reproductive age. Thus, the medical records of patients found to be carriers of any genetic disorder should be flagged, and individuals from affected racial/ethnic groups should be routinely advised to review their own and their partner’s genetic status as part of the family planning process. Parents who fail to consider carrier status may have children who inherit SCD, thalassemia, or a combination of these diseases.21,22
Porphyria
Porphyria may encompass an even wider spectrum of disorders than thalassemia. The various clinical entities have little in common other than their pathophysiology—disordered heme synthesis. As heme is synthesized, it transitions through several highly reactive states, and deficiencies in different areas result in different syndromes. When there is a deficiency in one of the enzymes in the heme degradation pathway, reactive metabolites upstream of the defect may occur.
However, all porphyrias have one common feature: the accumulation of porphyrins or their precursors, which sometimes gives the urine a reddish color. A number of porphyrias are extremely rare—only 5 cases of dehydrogenase deficiency porphyria have been reported, for example, and the incidence of congenital erythropoietic porphyria is <1 in a million.23,24
Others occur a bit more frequently in particular ethnic groups. Variegate porphyria, a hepatic form of the disorder associated with acute attacks and photosensitivity, is particularly common among the white South African population, for example.25 Among Eastern Europeans, the incidence of porphyria cutanea tarda (PCT)—the most common porphyria—may be as high as 1 in 5000.25 The “tarda” in the name reflects the fact that, unlike other porphyrias, onset of PCT occurs later in life.26
Inheritance
Inheritance varies by condition. PCT is autosomal dominant, as are most porphyrias; acute intermittent porphyria, however, is autosomal recessive. Some cases of PCT are acquired, occurring as a result of exposure to environmental or infectious agents.27
Testing
A random urine porphobilinogen (PBG) is a useful screening test for porphyria, although checking urine for fluorescence may be the most readily available clinical examination. Further delineation using urine and fecal porphyrins is usually not readily available, and may require sending specimens to a specialized laboratory. “GeneReviews” (https://www.ncbi.nlm.nih.gov/sites/GeneTests/review?db=GeneTests) is a useful resource for locating such labs; the site also provides educational material about genetic diseases as well as information about specimen collection and billing.
Clinical presentation
Characterized by irritable and erratic behavior provoked by sunlight (with varying degrees of hirsutism) and ameliorated by ingesting fresh blood, porphyria could have been the inspiration for the vampire legends.28 Although symptoms vary from one type of porphyria to another, most affect either the nervous system or the skin. Some porphyrias, including PCT, are associated with a blistering, photosensitive rash.29
The acute porphyrias—a grouping of several variants, including acute intermittent porphyria and variegate porphyria—typically cause severe abdominal pain and neurologic symptoms, while erythropoietic porphyria patients may present with anemia, hypo- and hyperpigmentation of the skin, red urine, and reddish coloration of the teeth (FIGURE).29 Porphyrias are an often-overlooked cause of neuropathy, as well.30
FIGURE
Erythropoietic porphyria: Distinguishing characteristics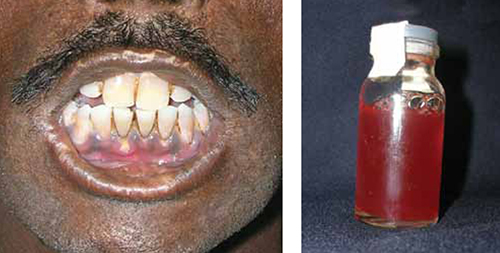
Patients with erythropoietic porphyria, like the one shown here, often present with reddish coloration of the teeth and red urine.
Treatment and follow-up
The abdominal pain associated with porphyria can be treated with dextrose infusions, analgesics, and hematin. Long-term management includes monitoring for cirrhosis, iron overload, and possibly, hepatocellular carcinoma.27
The treatment for anemia resulting from ineffective erythropoiesis—which ironically, results in iron overload—is phlebotomy, with 400 mL of blood removed every 2 weeks until the iron overload is relieved.31 Erythropoietin may be used to treat anemia resulting from phlebotomy, and is thought to mobilize iron stores.32