Helping patients with cystic fibrosis live longer






Lung and pancreas dysfunction are just part of the CF picture. Managing this “full-body disease” requires timely testing, traditional therapies, and familiarity with newer agents.
› Prescribe inhaled dornase alpha and inhaled tobramycin for maintenance pulmonary treatment of moderate to severe cystic fibrosis (CF). A
› Give aggressive nutritional supplementation to maintain a patient’s body mass index and blood sugar control and to attain maximal forced expiratory volume in one second (FEV1). B
› Consider prescribing cystic fibrosis transmembrane conductance regulator modulators, which have demonstrated a 5% to 10% improvement in FEV1 for CF patients. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence
B Inconsistent or limited-quality patient-oriented evidence
C Consensus, usual practice, opinion, disease-oriented evidence, case series
The focus of treatment. CF is not limited to the classic picture of lung and pancreas destruction with subsequent loss of function. The underlying pathology can occur in body epithelial tissues from the intestinal lining to sweat glands. These tissues contain cystic fibrosis transmembrane conductance regulator (CFTR), a protein that allows for the transport of chloride across epithelial cell membranes.2 In individuals homozygous for mutated CFTR genes, chloride transport can be impaired. In addition to regulating chloride transport, CFTR is part of a larger, complex interaction of ion transport proteins such as the epithelial sodium channel (ENaC) and others that regulate bicarbonate secretion.2 Decreased chloride ion transport in mutant CFTR negatively affects the ion transport complex; the result is a higher-than-normal viscosity of secreted body fluids.
Reason for hope. It is this impairment of chloride ion transport that leads to the classic phenotypic features of CF (eg, pulmonary function decline, pancreatic insufficiency, malnutrition, chronic respiratory infection), and is the target of both established and emerging therapies3—both of which I will review here.
When to consider a CF diagnosis
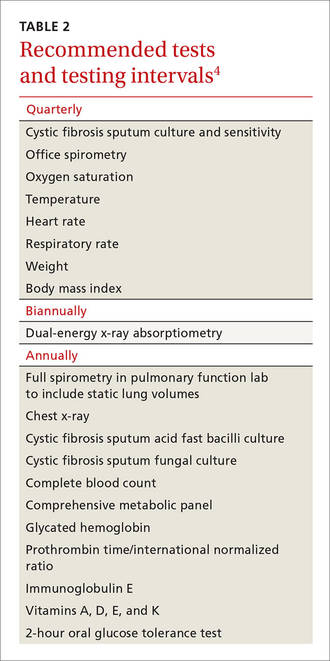
Cystic fibrosis remains a clinical diagnosis when evidence of at least one phenotypic feature of the disease (TABLE 1) exists in the presence of laboratory evidence of a CFTR abnormality.4 Confirmation of CFTR dysfunction is demonstrated by an abnormality on sweat testing or identification of a CF-causing mutation in each copy of CFTR (ie, one on each chromosome).5 All 50 states now have neonatal laboratory screening programs;4 despite this, 30% of cases in 2012 were still diagnosed in those older than 1 year of age, with 3% to 5% diagnosed after age 18.1
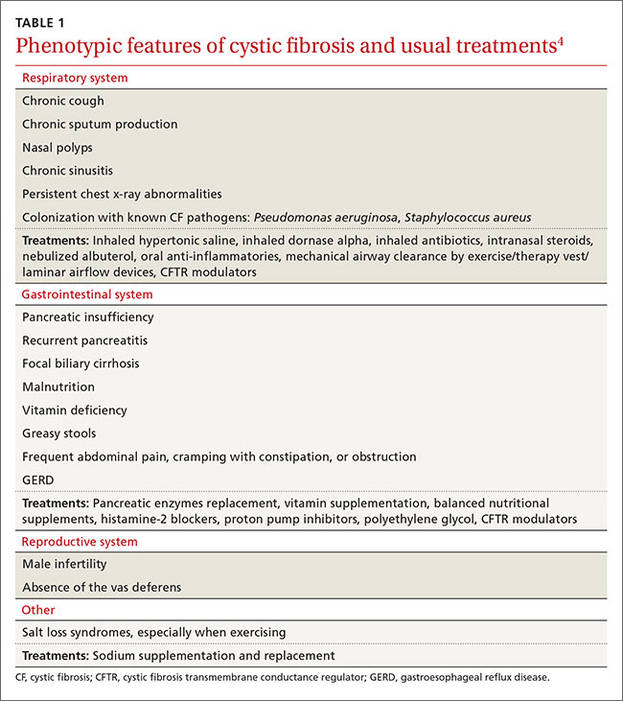
A sweat chloride reading in the abnormal range (>60 mmol/L) is present in 90% of patients diagnosed with CF in adulthood; this test remains the gold standard in the diagnosis of CF and the initial test of choice in suspected cases.4 Newborn screening programs identify those at risk by detecting persistent hypertrypsinogenemia and referring those with positive results for definitive testing with sweat chloride evaluation. Keep CF in mind when evaluating adolescents and adults who have chronic sinusitis, chronic/recurrent pulmonary infections, chronic/recurrent pancreatitis, or infertility from absence of the vas deferens.4 When features of the CF phenotype are present, especially if there is a known positive family history of CF or CF carrier status, order sweat chloride testing.
Traditional therapies
Both maintenance and acute therapies are directed throughout the body at decreasing fluid viscosity, clearing fluid with a high viscosity, or treating the tissue destruction that results from highly-viscous fluid.3 The traditional classic picture of CF is one of lung and pancreas destruction with subsequent loss of function. However, CF is, in reality, a full-body disease.
Respiratory system: Lungs
CFTR dysfunction in the lungs results in thick pulmonary secretions as the aqueous surface layer (ASL) lining the alveolar epithelium becomes dehydrated and creates a prime environment for the development of chronic infection. What ensues is a recurrent cycle of chronic infection, inflammation, and tissue destruction with loss of lung volume and function. Current therapies interrupt this cycle at multiple points.6
Airway clearance is one of the hallmarks of CF therapy, using both chemical and mechanical treatments. Daily, most patients will use either a therapy vest that administers sheering forces to the chest cavity or an airflow device that creates positive expiratory pressure and laminar flow to aid in expectorating pulmonary secretions.7 Because exercise has yielded comparable results to mechanical or airflow clearance devices, it is recommended that all CF patients who are not otherwise prohibited engage in regular, vigorous exercise in accordance with standard recommendations for the general public.7