Laugier-Hunziker Syndrome





Practice Points
- Laugier-Hunziker syndrome (LHS) comprises benign mucosal pigmentation in the absence of gastrointestinal pathology.
- Differentiating LHS from Peutz-Jeghers syndrome can prevent unnecessary aggressive cancer screening protocols.
- The average age of onset of LHS is 52 years and typically occurs in white adults.
- Pigmentation in LHS is most commonly distributed on the lower lips and oral mucosa.
Benign hyperpigmentation of the lips and fingers has been reported.1 The average age of onset of LHS is 52 years, and it typically is diagnosed in white adults.1,2 In LHS, pigmentation is most commonly distributed on the lips, especially the lower lips and oral mucosa.2 Pigmentation of the nails in the form of longitudinal melanonychia is present in approximately half of cases.2,3 There also may be pigmentation of the neck; thorax; abdomen; and acral surfaces, especially the fingertips.1-3 Rarely, pigmented macules can occur on the genitalia or sclera.1,2 Unlike Peutz-Jeghers syndrome, the diagnosis of LHS does not result from a germline mutation and carries no risk of gastrointestinal polyposis or internal malignancy.3,4 The histopathology of a pigmented macule of LHS shows a normal number and morphology of melanocytes. Epidermal basement membrane pigmentation is common, with pigment-laden macrophages evident in the papillary dermis.3
RELATED ARTICLE: Asymptomatic Lower Lip Hyperpigmentation From Laugier-Hunziker Syndrome
The differential diagnosis of multiple lentigines is broad and includes Peutz-Jeghers syndrome; LEOPARD (lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) syndrome; Carney complexes, including LAMB (lentigines, atrial myxoma, mucocutaneous myxoma, blue nevi) and NAME (nevi, atrial myxoma, myxoid neurofibroma, ephelide) syndromes5; primary adrenocortical insufficiency (Addison disease); and idiopathic melanoplakia.2 Peutz-Jeghers syndrome, an autosomal-dominant syndrome with mucocutaneous lentigines, has a similar clinical appearance to LHS; therefore, it is necessary to exclude this diagnosis due to its association with intestinal hamartomatous polyps and internal malignancies (Table).3,6,7
,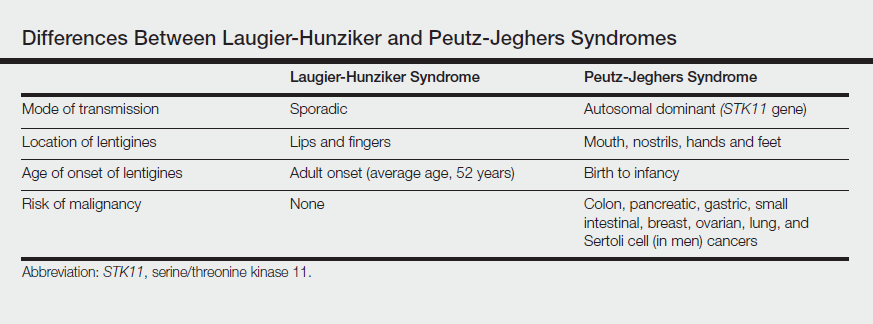
Peutz-Jeghers syndrome is characterized by mucocutaneous hyperpigmentation and intestinal hamartomatous polyposis and is associated with internal malignancies of the colon, breast, pancreas, stomach, small intestines, ovaries, lung, and Sertoli cells in men.6,7 Associated gastrointestinal tract malignancies in descending order of frequency are colon (39%), pancreatic (36%), gastric (29%), and small intestine (13%).1 It is caused by a germ line mutation of the serine/threonine kinase 11 gene, STK11. Although the appearance and distribution of the mucocutaneous lentigines is similar to individuals with LHS, by contrast the lentiginosis in individuals with Peutz-Jeghers syndrome is present from birth or develops during infancy.6 Aggressive cancer screening guidelines aid in early detection and begin at 8 years of age with a baseline colonoscopy and esophagogastroduodenoscopy; future screening is dictated by the presence or absence of polyps. If no polyps are detected at 8 years of age, a colonoscopy and esophagogastroduodenoscopy are repeated at 18 years of age and then every 3 years until 50 years of age.8
In an adult patient, the diagnosis of LHS can be made clinically and a correct diagnosis prevents frequent and unpleasant gastrointestinal tract cancer screening examinations. Lampe et al2 described a man with LHS who was incorrectly diagnosed with Peutz-Jeghers syndrome and experienced a colonic perforation as a complication of a screening colonoscopy. Their case report underscores the importance of making the correct diagnosis of LHS to avoid undertaking unnecessary aggressive cancer screening regimens.2
Although LHS is a benign condition that does not require treatment, Q-switched alexandrite or erbium:YAG laser therapy has been shown to improve the pigmentary findings associated with LHS.9,10 It has been suggested that LHS should be renamed Laugier-Hunziker pigmentation2 or mucocutaneous lentiginosis of Laugier and Hunziker1 to differentiate LHS as simply a disorder of pigmentation rather than a potentially morbid genetic defect, as in Peutz-Jeghers syndrome.