A Solitary Axillary Subcutaneous Mass





THE DIAGNOSIS: Cutaneous Rosai-Dorfman Disease
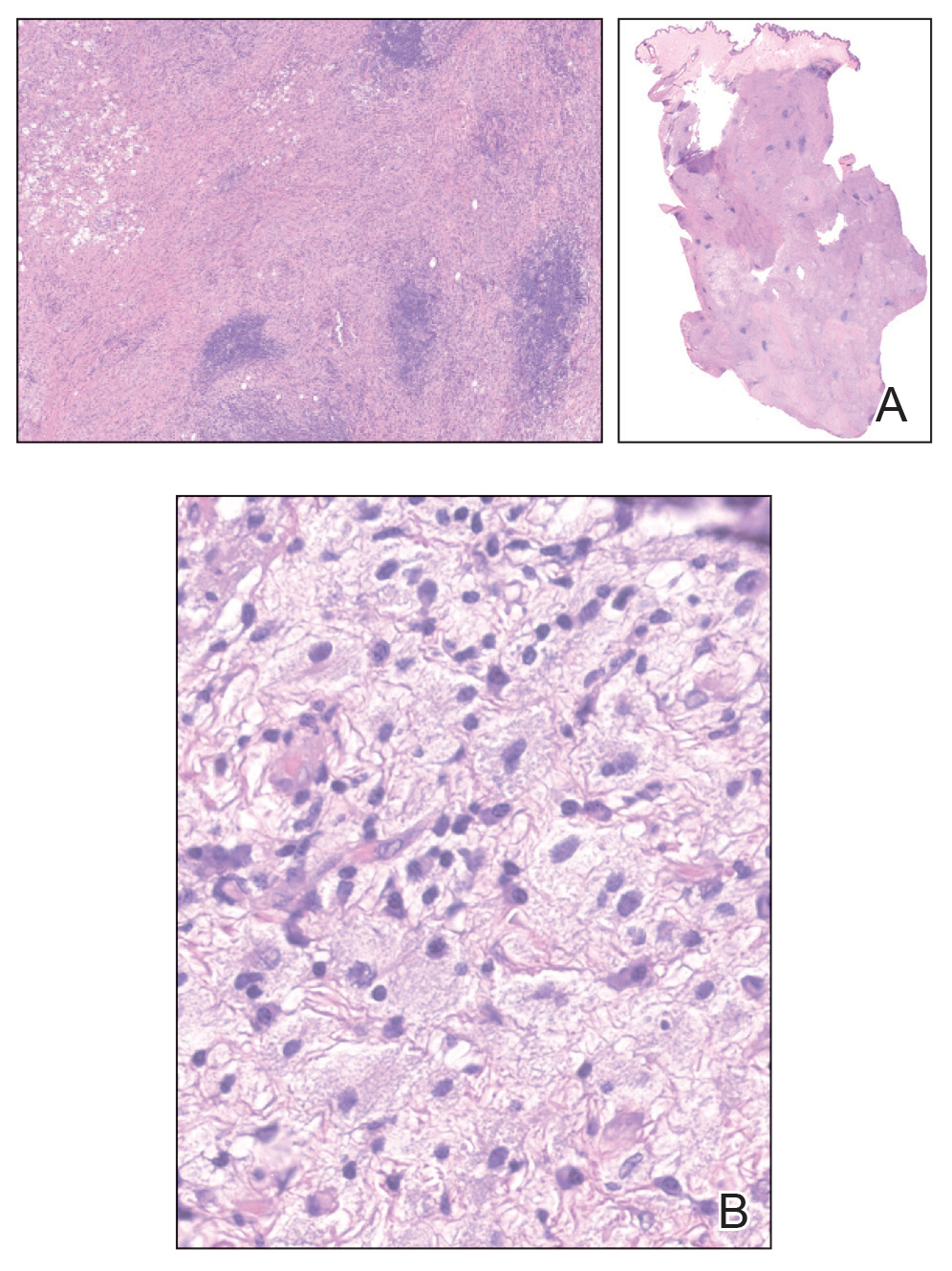
The clinical differential diagnosis in our patient included a broad array of soft-tissue neoplasms ranging from benign entities to sarcomas. Histology was notable for a dense, dermal-based, lymphohistiocytic infiltrate with alternating hypocellular and hypercellular areas imparting a marbled appearance on low-power view (Figure, A). Further immunohistochemical staining revealed large, S100-positive histiocytes containing intact inflammatory cells (emperipolesis), which confirmed a diagnosis of cutaneous Rosai-Dorfman disease (RDD)(Figure, B). Our patient elected to undergo surgical removal of the mass, and he will be monitored for recurrence.
Rosai-Dorfman disease is a non–Langerhans cell histiocytosis that most commonly affects the lymph nodes but can affect other organs including the skin. Rosai-Dorfman disease initially was documented in the medical literature in 1969 by Rosai and Dorfman1 as benign sinus histiocytosis with massive lymphadenopathy. Classic RDD usually manifests with painless cervical lymphadenopathy in children or young adults along with fever, leukocytosis, anemia, polyclonal hypergammaglobulinemia, and elevated inflammatory markers.2,3 Extranodal involvement has been reported in up to 43% of cases, with common sites including the skin, central nervous system, and gastrointestinal tract.3,4
Cutaneous RDD is a distinct, less common clinical entity that is limited to the skin and shows no nodal involvement or systemic symptoms such as fever, night sweats, or weight loss.5 Cutaneous RDD classically manifests with localized indurated papules and plaques, but it can manifest with tumorlike lesions in the subcutaneous tissues.6 Cutaneous RDD is very rare, with fewer than 200 known case reports in the literature as of 2014; in comparison to classic forms of RDD, cutaneous RDD has a female predominance.7,8 There are few reports of isolated cutaneous disease manifesting as soft-tissue masses, and our case represents a rare case of cutaneous RDD manifesting as a solitary soft-tissue mass in the axilla.9-11 Diagnosis of cutaneous RDD is challenging due to its variable clinical manifestations and nonspecific imaging findings, requiring clinicopathologic correlation.
Imaging of subcutaneous RDD lesions typically shows well-defined, irregularly shaped masses with homogenous enhancement on computed tomography/ magnetic resonance imaging. Additional imaging with positron emission tomography/computed tomography is recommended to examine for organ involvement, as RDD lesions have avid uptake.12,13 Imaging may help differentiate RDD lesions from malignant neoplasms prior to biopsy. Additional workup includes baseline laboratory testing with inflammatory markers and a complete blood count for evaluation of laboratory abnormalities seen in classic RDD, including leukocytosis, anemia, or systemic inflammation.12 Following imaging and laboratory testing, definitive diagnosis of RDD necessitates histopathologic examination.
Although cutaneous RDD is clinically distinct from its classic RDD counterpart, the conditions share the same characteristic histologic features.5 Histology is notable for a dense mixed inflammatory infiltrate comprised of large pale histiocytes exhibiting emperipolesis, lymphocytes, plasma cells, and occasional eosinophils and neutrophils. Histiocytes stain positive for CD68, CD163, and S100 and are negative for Langerhans cell markers CD1a and CD207.6
The etiology of RDD remains poorly understood. Classic RDD has been associated with both sporadic and familial forms, with somatic mutations identified in the mitogen-activated protein kinase/KRAS pathway in up to one-third of cases, and less frequently in the BRAF gene.14,15 Germline mutations in familial cases of RDD have been identified in the SLC29A3 gene; mutations in this gene are associated with a spectrum of syndromes with histiocytosis and lymphadenopathy.14,15 In contrast, molecular drivers have yet to be identified in cutaneous RDD lesions, and the current predominant hypothesis is that cutaneous RDD has a reactive or immunologic pathophysiology. Autoimmune diseases, infections, and lymphomas have been reported to co-occur with both classic and cutaneous RDD.15 While subclinical viral infections such as Epstein-Barr virus and human herpesvirus 6 have been identified in RDD cases, studies have failed to prove their role as pathogenic drivers of the disease.14,16,17 Commonly reported comorbidities include systemic lupus erythematous, diabetes, hemolytic anemia, acute/chronic uveitis (though it is controversial whether these cases represent orbital involvement in systemic RDD), and Crohn disease.7,8,18,19 Immunohistochemical findings have supported that cells within RDD are activated monocytes responding to T-cell cytokine signaling following an infectious or immunologic insult.20,21
Consensus guidelines on treatment for cutaneous RDD recommend either observation for asymptomatic disease or surgical excision for unifocal lesions with consideration of systemic therapy for refractory cutaneous disease.22,23 Most patients with cutaneous RDD have self-limited disease, but long-term follow-up is recommended following surgical excision to monitor for recurrence, especially if there is a residual positive margin.24 Radiation therapy also may have to be utilized for residual or recurrent disease that becomes symptomatic; however, further studies are needed to determine its efficacy in limiting recurrence.4,12,25 Systemic treatment options include immunosuppressive or immunomodulatory agents such as corticosteroids, methotrexate, and rituximab.5 There currently are no guidelines on length of follow-up, but surveillance is recommended initially at 4 months, followed by 6- to 12-month intervals.22