Antisynthetase syndrome: Not just an inflammatory myopathy





ABSTRACTIn recent years, antisynthetase syndrome has been recognized as an important cause of autoimmune inflammatory myopathy in a subset of patients with polymyositis and dermatomyositis. It is associated with serum antibodies to aminoacyl-transfer RNA synthetases and is characterized by a constellation of manifestations, including fever, myositis, interstitial lung disease, “mechanic’s hands,” Raynaud phenomenon, and polyarthritis. Physicians should be familiar with its variety of clinical presentations and should include it in the differential diagnosis in patients presenting with unexplained interstitial lung disease.
KEY POINTS
- Antisynthetase syndrome can present with a wide variety of clinical manifestations, including myositis and interstitial lung disease.
- The type and severity of interstitial lung disease usually determine the long-term outcome.
- In the appropriate clinical setting, the diagnosis is usually confirmed by the detection of antibodies to various aminoacyl-transfer RNA synthetases, anti-Jo-1 antibody being the most common.
- Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine or methotrexate are often required as steroid-sparing agents and also to achieve disease control.
- In the case of severe pulmonary involvement, cyclophosphamide is recommended.
RARE BUT UNDERRECOGNIZED
The true population prevalence of antisynthetase syndrome is unknown. Because this syndrome is rare, comprehensive epidemiologic studies are difficult to perform.
In several retrospective studies, the annual incidence of idiopathic inflammatory myopathies has been reported to be 2 to 10 new cases per million adults per year.3 Antisynthetase antibodies are detected in 20% to 40% of such cases.4–6 The disease is two to three times more common in women than in men.7
Early diagnosis is difficult because the clinical presentation is varied and often nonspecific, clinically milder disease may escape detection, and many general practitioners lack familiarity with this syndrome and consequently do not recognize it. Moreover, tests for myositis-specific antibodies (including antisynthetase antibodies) are often not ordered in the evaluation of myositis, and hence the diagnosis of antisynthetase syndrome cannot be substantiated. Furthermore, interstitial lung disease can predominate or can be the sole manifestation in the absence of clinically apparent myositis,8–10 and patients can be misdiagnosed as having idiopathic pulmonary fibrosis when underlying antisynthetase syndrome is not suspected. This distinction may be important because these conditions differ in their pathology and treatment. Histologically, the predominant pattern of lung injury in idiopathic pulmonary fibrosis is “usual interstitial pneumonia” which does not respond to immunosuppressive therapy, and hence lung transplantation is the only therapeutic option. On the other hand, in antisynthetase syndrome, the usual pattern of lung injury is “nonspecific interstitial pneumonia,” in which immunosuppressive therapy has a role.
Anti-Jo-1 antibody is detected in 15% to 25% of patients with polymyositis and in up to 70% of myositis patients with concomitant interstitial lung disease.11 Autoantibodies to seven other, less frequently targeted, aminoacyl tRNA synthetases have also been described in patients with polymyositis and interstitial lung disease (Table 1).11,12 In addition, an autoantibody to a 48-kDa transfer RNA-related protein (Wa) has been described.13 These non-Jo-1 antisynthetase antibodies are detected in only about 3% of myositis patients.14
ROLE OF ANTISYNTHETASE ANTIBODIES
Synthetases play a central role in protein synthesis by catalyzing the acetylation of tRNAs. The propensity of organ involvement in antisynthetase syndrome suggests that tissue-specific changes in muscle or lung lead to the production of unique forms of target autoantigens, the aminoacyl-tRNA synthetases. There is evidence that these enzymes themselves may be involved in recruiting both antigen-presenting and inflammatory cells to the site of muscle or lung injury.15 However, the molecular pathway that initiates and propagates this autoimmune response and the specific role of the antisynthetase antibodies in the pathogenesis of this syndrome are presently unknown.
SIX SALIENT CLINICAL FEATURES
There are six predominant clinical manifestations, which may be present at disease onset or appear later as the disease progresses:
- Fever
- Myositis
- Interstitial lung disease
- Mechanic’s hands
- Raynaud phenomenon
- Inflammatory polyarthritis.
There is considerable clinical heterogeneity, and one or other manifestation can predominate or can be the only expression of the syndrome. Furthermore, in the same patient, the individual features can prevail at different times and may develop years after onset of the disease. Therefore, in addition to patients with myositis, it would be important to suspect antisynthetase syndrome in patients presenting with isolated lung involvement (amyopathic interstitial lung disease), as there are therapeutic implications. Studies have demonstrated the efficacy of immunosuppressive agents in interstitial lung disease associated with antisynthetase syndrome (where the predominant pattern of lung injury is “nonspecific interstitial pneumonitis”), whereas lung transplantation has so far been the only treatment option in idiopathic pulmonary fibrosis.
Fever
About 20% of patients have a fever at disease onset or associated with relapses. Sometimes the fever can persist until treatment of antisynthetase syndrome is started.
Myositis
Muscle disease is seen in more than 90% of patients with anti-Jo-1 antisynthetase syndrome. It can be subclinical (in the absence of proximal myopathy), manifested by transient creatine kinase elevation only, which may normalize after therapy is initiated.
However, more commonly, patients develop profound proximal muscle weakness and sometimes muscle pain (Table 2). Weakness of the striated muscles of the upper esophagus, cricopharyngeus, and hypopharynx may cause dysphagia and makes these patients susceptible to aspiration pneumonia. Diaphragmatic and intercostal muscle weakness can contribute to shortness of breath in some patients. Myocarditis has also been reported.
Pulmonary disease
Interstitial lung disease develops in most patients with anti-Jo-1 antisynthetase syndrome, with a reported prevalence of about 90% in one series.16 Patients often present with acute, subacute, or insidious onset of exertional dyspnea. Sometimes there is an intractable nonproductive cough.
At the outset of antisynthetase syndrome, if the patient is profoundly weak because of myopathy or has inflammatory polyarthritis, mobility is significantly compromised, and exertional dyspnea may not be experienced. However, as the interstitial lung disease progresses, shortness of breath becomes overt, more so when the patient’s level of activity improves with treatment of myositis.
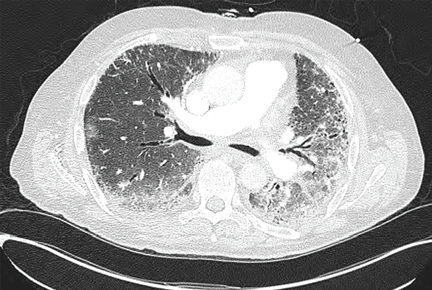
Inspiratory crackles on auscultation of the lung bases or changes on chest radiography are relatively insensitive findings and can miss early interstitial lung disease. Therefore, if antisynthetase syndrome is suspected or diagnosed, a baseline pulmonary function test (spirometry and carbon monoxide diffusion capacity) is indicated. It will often detect occult interstitial lung disease, and the diagnosis can then be confirmed with thoracic high-resolution computed tomography (Figure 2).
Pulmonary hypertension. Recent studies indicate that, similar to patients with other autoimmune rheumatic diseases, pulmonary hypertension can develop in patients with antisynthetase syndrome, with or without concomitant interstitial lung disease.17,18 This complication occurred in the case presented here. It has been found that when pulmonary hypertension coexists with interstitial lung disease, its degree may not correlate with the severity of the latter.17 Additionally, pulmonary hypertension, when present, has been found to contribute independently to prognosis and survival.