Antisynthetase syndrome: Not just an inflammatory myopathy





ABSTRACTIn recent years, antisynthetase syndrome has been recognized as an important cause of autoimmune inflammatory myopathy in a subset of patients with polymyositis and dermatomyositis. It is associated with serum antibodies to aminoacyl-transfer RNA synthetases and is characterized by a constellation of manifestations, including fever, myositis, interstitial lung disease, “mechanic’s hands,” Raynaud phenomenon, and polyarthritis. Physicians should be familiar with its variety of clinical presentations and should include it in the differential diagnosis in patients presenting with unexplained interstitial lung disease.
KEY POINTS
- Antisynthetase syndrome can present with a wide variety of clinical manifestations, including myositis and interstitial lung disease.
- The type and severity of interstitial lung disease usually determine the long-term outcome.
- In the appropriate clinical setting, the diagnosis is usually confirmed by the detection of antibodies to various aminoacyl-transfer RNA synthetases, anti-Jo-1 antibody being the most common.
- Although glucocorticoids are considered the mainstay of treatment, additional immunosuppressive agents such as azathioprine or methotrexate are often required as steroid-sparing agents and also to achieve disease control.
- In the case of severe pulmonary involvement, cyclophosphamide is recommended.
A 66-year-old man was initially seen in clinic in March 2004 with a 5-month history of polyarthritis (affecting the finger joints, wrists, and knees) and several hours of morning stiffness. He also had significant proximal muscle weakness, progressive exertional dyspnea, and a nonproductive cough. There was no history of fever, chills, rash, dysphagia, or sicca symptoms. Findings on initial tests:
- His creatine kinase level was 700 U/L (reference range 30–220), which later rose to 1,664 U/L.
- He was positive for antinuclear antibody with a 5.7 optical density ratio (normal < 1.5) and for anti-Jo-1 antibody.
- An electromyogram was consistent with a necrotizing myopathy. Left rectus femoris biopsy revealed scattered degenerating and regenerating muscle fibers but no evidence of endomysial inflammation.
- On pulmonary function testing, his forced vital capacity was 80% of predicted, and his carbon monoxide diffusion capacity was 67% of predicted.
- High-resolution computed tomography revealed evidence of interstitial lung disease, characterized by bilateral patchy ground-glass opacities suggestive of active alveolitis, most extensive at the lung bases.
- Bronchoalveolar lavage indicated alveolitis, and transbronchial biopsy revealed pathologic changes consistent with cryptogenic organizing pneumonia. All cultures were negative.
This constellation of clinical manifestations, including myositis, interstitial lung disease, and polyarthritis, along with positive anti-Jo-1 antibody, confirmed the diagnosis of antisynthetase syndrome.
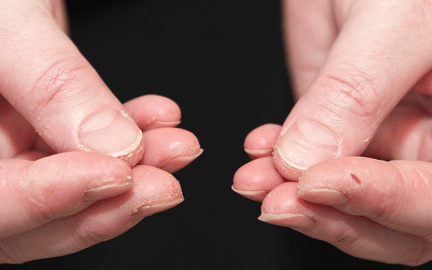
In June 2004, for his interstitial lung disease, he was started on daily oral cyclophosphamide along with high-dose oral prednisone. Three months later the skin of the tips and radial margins of his fingers started thickening and cracking, the appearance of which is classically described as “mechanic’s hands,” a well-described manifestation of antisynthetase syndrome (Figure 1).
Cyclophosphamide was continued for about a year. Subsequently, along with prednisone, he sequentially received various other immunosuppressive medications (methotrexate, tacrolimus, mycophenolate mofetil, and rituximab) over the next few years in an attempt to control his progressive interstitial lung disease. All of these agents were only partially and temporarily effective. Ultimately, despite all of these therapies, as his interstitial lung disease progressed, he needed supplemental oxygen and enrollment in a pulmonary rehabilitation program.
In March 2010, he was admitted with worsening dyspnea and significant peripheral edema and was found to have severe pulmonary arterial hypertension. He was started on bosentan. Eight months later sildenafil was added for progressive pulmonary arterial hypertension. However, his oxygenation status continued to decline.
In July 2011, he presented with chills, increasing shortness of breath, and a mild productive cough. As he was severely hypoxic, he was admitted to the intensive care unit and started on mechanical ventilation and broad-spectrum antibiotics. Despite escalation of oxygen therapy, his respiratory status rapidly deteriorated, and he developed hypotension requiring vasopressors. He ultimately died of cardiac arrest secondary to respiratory failure.
A CONSTELLATION OF MANIFESTATIONS
Antisynthetase syndrome, associated with anti-aminoacyl-transfer RNA (tRNA) synthetase antibodies, is characterized by a constellation of manifestations that include myositis, interstitial lung disease, mechanic’s hands, fever, Raynaud phenomenon, and nonerosive symmetric polyarthritis of the small joints.1
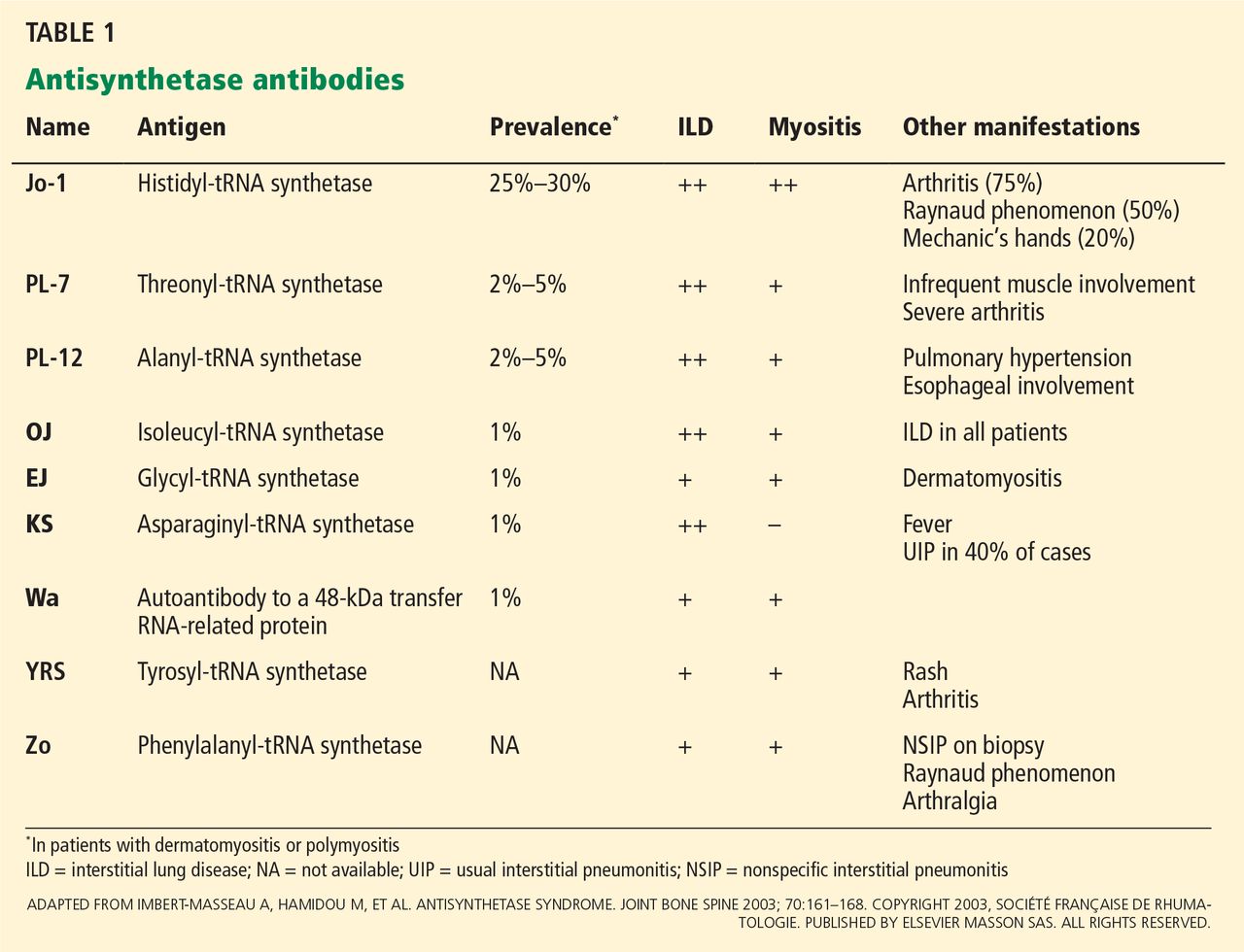
Anti-Jo-1 antibody (anti-histidyl-tRNA synthetase) is the most common of the antibodies and also was the first one to be identified (Table 1). It was named after John P, a patient with polymyositis and interstitial lung disease, in whom it was first detected in 1980.2 The onset of the syndrome associated with anti-Jo-1 antibody is often acute, and the myositis is usually steroid-responsive. However, not uncommonly, severe disease can develop over time, with a tendency to relapse and with a poor long-term prognosis.