A Howling Cause of Pancytopenia





The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similar to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
© 2018 Society of Hospital Medicine
COMMENTARY
Because the differential diagnosis for new-onset pancytopenia encompasses many diseases across several medical subspecialties, a thorough history and physical exam are necessary to form a tailored clinical approach.1 The primary causes of pediatric pancytopenia vary depending on geographic location because of the local prevalence of infectious agents and nutritional deficiency patterns. A retrospective study investigating the primary cause of pancytopenia in children without existing malignancy presenting to a US tertiary care hospital found that 64% of cases were due to infection, 28% were due to hematologic disease (most frequently aplastic anemia), and 8% were due to miscellaneous etiologies, including adverse drug reactions and autoimmune diseases.2 In contrast, the most common cause of pancytopenia in pediatric patients presenting to a tertiary care hospital in India was megaloblastic anemia (28%), followed by infections (21%), acute leukemia (21%), and aplastic anemia (20%).3 While clinicians do (and should) consider malignancy as a cause of pancytopenia, there is sparse literature regarding the frequency of pancytopenia associated with the presentations of childhood malignancies.4 A study of pediatric patients with acute lymphoblastic anemia found that only 11% of newly diagnosed patients had pancytopenia at initial presentation.4
There are no official guidelines for the work-up of pediatric pancytopenia from any of the academic societies. Depending on the clinical history, initial laboratory investigation for pediatric pancytopenia may include complete blood cell count with differential, reticulocyte count, peripheral blood smear, complete metabolic panel, hemolysis labs (haptoglobin, LDH, Coombs test) and inflammatory markers (ESR, CRP, fibrinogen). Further investigation to clarify the specific etiology of pancytopenia can be guided by the results of these initial tests.
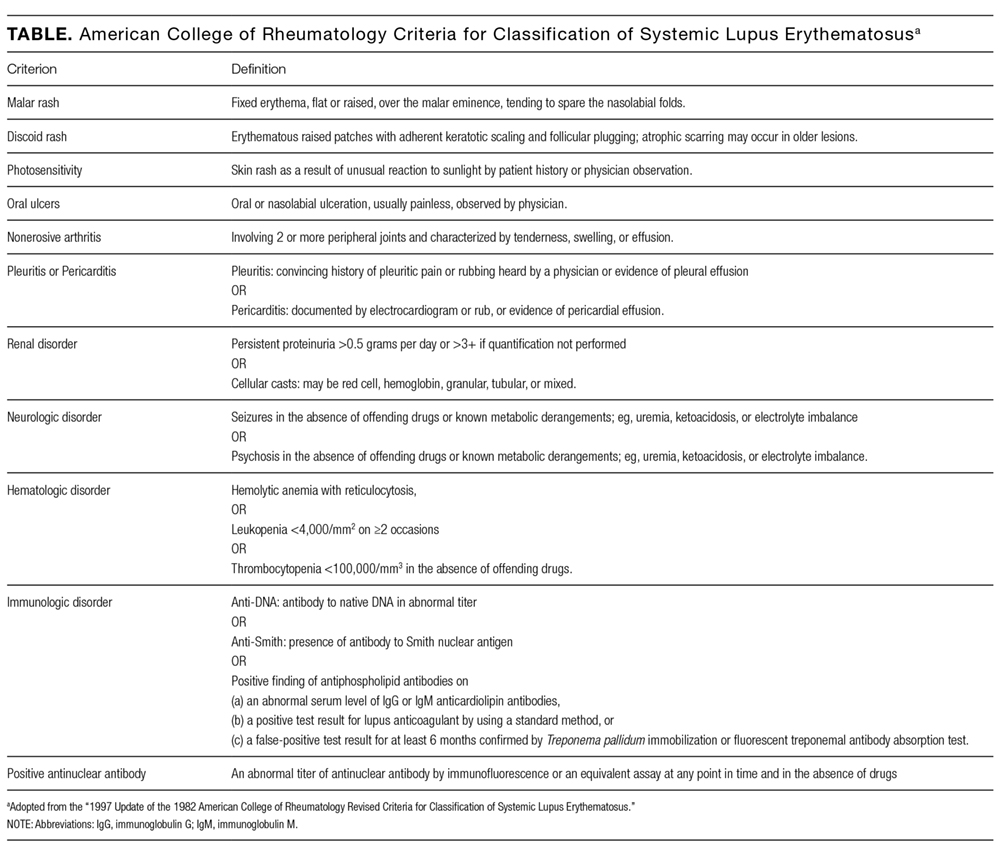
Hematologic abnormalities are present in greater than 70% of pediatric SLE cases.10,11 The pathogenesis of hematologic abnormalities in SLE is heterogeneous, involving actions of autoreactive lymphocytes, autoantibodies, and proinflammatory cytokines that can disrupt bone marrow production and cause peripheral blood cell destruction.12,13 While pancytopenia is common in children with SLE, other coexisting diagnoses should be considered in patients with SLE and pancytopenia. Concurrent diagnoses that can lead to pancytopenia in patients with SLE include infection, pharmacologic side effects, and secondary HLH,14,15 each of which has differing implications for prognosis and treatment.
Secondary HLH is a severe and often acute complication of systemic inflammatory disorders caused by the proliferation and activation of T cells and macrophages, leading to an enhanced inflammatory state. When HLH occurs in the setting of an underlying autoimmune or autoinflammatory process, it is typically termed MAS. MAS affects an estimated 0.9% to 4.6% of patients with SLE.16 Early diagnosis and treatment of MAS is important because MAS can be rapidly fatal, with a mortality rate of 8% to 20% in pediatric patients.17,18 Clinical features of MAS include physical exam findings of fever and splenomegaly as well as laboratory abnormalities, including pancytopenia, elevated ferritin, elevated triglycerides, and low fibrinogen.18 A bone marrow biopsy showing hemophagocytosis in the absence of malignancy is diagnostic of MAS. Although a bone marrow biopsy is not required to diagnose MAS, it is often obtained to exclude other etiologies of pancytopenia such as malignancy.19 Specialized diagnostic testing for MAS includes NK cell counts and functional studies, including expression of perforin and granzyme B (NK cell proteins triggering apoptosis in target cells), soluble IL-2R (marker of activated lymphocytes), and CD163 (transmembrane protein of hemophagocytic macrophages). There is no standardized protocol for treating MAS.20 It is most commonly treated with highdose corticosteroids; additional agents, including cyclosporine and biologic therapies, are also utilized.16,20