A Howling Cause of Pancytopenia





The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similar to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
© 2018 Society of Hospital Medicine

The pancytopenia is obviously notable. It raises the possibility that the oral ulcerations are due to the neutropenia rather than a primary disease manifestation. Other possible causes of pancytopenia include SLE, antiphospholipid antibody syndrome, and related rheumatologic diagnoses, including hemophagocytic lymphohistiocytosis (HLH). Given her age and subacute presentation, secondary forms of HLH seem more likely than primary (genetic) forms, which typically present within the first few years of life. Secondary forms of HLH can occur in association with rheumatic diseases and are then referred to as Macrophage Activation Syndrome (MAS). The most common rheumatologic diseases associated with MAS are systemic juvenile idiopathic arthritis, SLE, and Kawasaki disease. Secondary HLH can also occur with infectious diseases, particularly viral infections such as EBV. It is also important to consider thrombotic thrombocytopenic purpura and other forms of thrombotic microangiopathy, especially if her violaceous eyelids actually represent purpura. The presence of pancytopenia also expands the differential diagnosis to include leukemia, lymphoma, and other oncologic diseases. After obtaining results from pending infectious disease studies, additional diagnostic work-up should include examination of the bone marrow and a peripheral blood smear to evaluate for hemophagocytosis and/or malignancy. Testing for double-stranded DNA antibodies and antinuclear antibodies (ANA) should be sent to evaluate for SLE, and antiphospholipid antibodies should also be checked. Renal function must also be evaluated.
Additional laboratory work-up revealed a reticulocyte count of 0.2%, a positive Coombs immunoglobulin G (IgG) test, haptoglobin less than 80 mg/L, and lactate dehydrogenase (LDH) 25.2 µkat/L (1509 units/L); coagulation studies were normal. Her chemistries showed electrolytes, blood urea nitrogen, and creatinine were within normal limits; her aspartate aminotransferase was 216 units/L, and alanine aminotransferase was 56 units/L. Her spot urine protein-to-creatinine ratio was 1.28. Complement and inflammatory studies showed C3 0.14 g/L (14 mg/dL, normal 83-151 mg/dL), C4 0.05 g/L (5 mg/dL, normal 13-37 mg/dL), erythrocyte sedimentation rate (ESR) 103 mm/hr (normal 0-20 mm/hr), and C-reactive protein (CRP) 3.2 mg/L (normal 0.7-1.7 mg/L). Additional studies showed elevated triglycerides (376 mg/dL), elevated creatine kinase (2437 units/L), and elevated ferritin (22,295.5 ng/mL). An ANA screen and specific autoantibody studies were sent, including antidouble stranded DNA antibody, antiribonucleoprotein antibody, anti-Smith antibody, anti-Ro antibody, and anti-La antibody. A bone marrow biopsy was performed.
The hematologic studies provide a mixed picture. There is evidence of an autoimmune hemolytic anemia (AIHA). Typically, AIHA is associated with reticulocytosis rather than reticulocytopenia. Reticulocytopenia can occur in AIHA, however, because of antibodies directed against erythroid precursors or if 2 processes are occurring simultaneously—ie, AIHA plus bone marrow destructive/failure process. The latter scenario is more likely here. Specifically, the pancytopenia, elevated triglycerides, and extreme hyperferritinemia strongly support the diagnosis of HLH. The very low C3 and C4 suggest a complement-consumptive process, and SLE is the most likely etiology. Proteinuria and Coombs-positive anemia are also features of SLE. The discordance between the ESR (markedly elevated) and CRP (mild elevation) is surprising in the setting of systemic inflammation. However, her other clinical features are consistent with marked systemic inflammation, and it is important not to dismiss a likely diagnosis simply on the basis of a few incongruous features. At this point, the diagnosis of SLE complicated by secondary HLH is favored, remembering that both these entities can be triggered by a viral infection. Therefore, diligent follow-up of the aforementioned specific autoantibody studies and the bone marrow biopsy is the next logical step, along with the still-pending infectious disease studies.
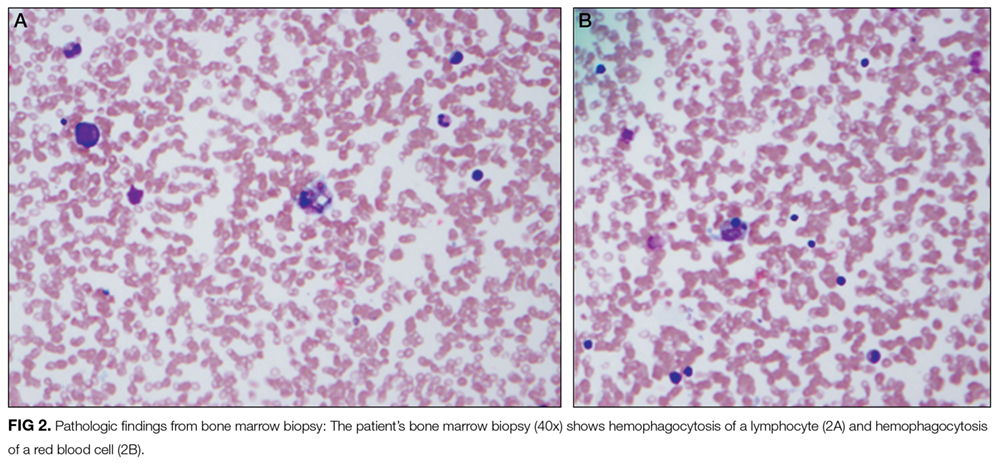
These findings are consistent with the diagnosis of SLE complicated by secondary HLH (ie, MAS). It remains possible, but unlikely, that the patient has genetic or familial HLH (fHLH), as this entity is exceedingly rare with distinct underlying genetic aberrations separate from SLE. Ideally, the NK cell function studies would be repeated after the current episode of HLH is controlled and the patient is off of immunosuppressive therapies, but this will likely not be possible given the underlying SLE. Patients with fHLH have reduced or absent NK cell function at baseline (ie, not only during an acute episode of HLH and not because of immunosuppressive medications). Alternatively, one could consider genetic testing for fHLH. The clinical importance of doing this is that patients with fHLH are candidates for bone marrow or stem cell transplantation. There currently is not a published standard of care for the work-up and management of MAS in children with rheumatic disease, so the decision to repeat NK cell function testing and/or genetic testing would be left to the discretion of the treating physician and would depend on the patient’s ongoing clinical course.
The patient required red blood cell and platelet transfusions. She received pulse dose intravenous methylprednisolone for treatment of SLE and MAS; she clinically improved within 48 hours of starting steroids. Cyclosporine was added for management of MAS. The patient was transitioned to oral corticosteroids and discharged home. All cell counts normalized within 1 month of discharge. She was weaned off corticosteroids and cyclosporine was discontinued. Her maintenance SLE therapy includes hydroxychloroquine and mycophenolate mofetil.