Mass Confusion





© 2017 Society of Hospital Medicine
Sporadic CJD, which accounts for approximately 85% of all cases of prion disease in humans, typically manifests with rapidly progressive dementia and myoclonus after a prolonged incubation period in persons between 55 and 75 years of age. Genetic forms account for approximately 15% and acquired forms less than 1% of human prion diseases.1 Prion diseases have a broad spectrum of clinical manifestations, including dementia, ataxia, parkinsonism, myoclonus, insomnia, paresthesias, and abnormal or changed behavior.4 Given the protean clinical manifestations of prion diseases and rarity, the diagnosis is challenging to make antemortem. One recent study showed that most patients receive about 4 misdiagnoses and are often two-thirds of the way through their disease course before the correct diagnosis of sporadic CJD is made.5
Testing for protein markers of rapid neuronal injury8 in the CSF including 14-3-3, total tau, and neuron-specific enolase can increase suspicion for CJD, although there is a 10%-50% false positive rate with these markers.9 In this case, those tests were not performed; positive results would have been even more nonspecific in the setting of an enhancing brain mass and recent brain surgery.
Although not available at the time this patient was evaluated, the real-time quaking-induced conversion (RT-QuIC) test performed in CSF is diagnostically helpful, and, if positive, supportive of the MRI findings. The sensitivity and specificity of this test have been reported to be between 87%-91% and 98%-100%, respectively, albeit with limited data.10 Applying RT-QuIC to nasal mucosal brushings might lead to even higher sensitivity and specificity.11Seeking a premortem diagnosis for a rare disease with no known cure may seem superfluous, but it has important implications for establishing prognosis, limiting subsequent diagnostic and therapeutic measures, and safeguarding of other patients and operating room personnel. Iatrogenic CJD has occurred following invasive procedures involving neurosurgical instrumentation.12 CJD has been transmitted from grafts of dura mater, transplanted corneas, implantation of inadequately sterilized electrodes in the brain, and in the early 1980s, injections of contaminated pituitary hormones (particularly growth hormone) derived from human pituitary glands taken from cadavers. Since CJD was first described in the 1920s, less than 1% of human prion cases have been acquired iatrogenically.13In patients with rapidly progressive cognitive decline who warrant brain biopsy or surgery, the probability of prion diseases should be assessed based on clinical information and the results of MRI, EEG, and CSF testing. If prion disease is plausible, World Health Organization14 precautions should be employed for neuroinvasive procedures to reduce transmission risk. Disposable equipment should be used when possible, and nondisposable neurosurgical instruments should be quarantined until a nonprion disease diagnosis is identified, or should be regarded as contaminated and reprocessed using the aforementioned protocol.
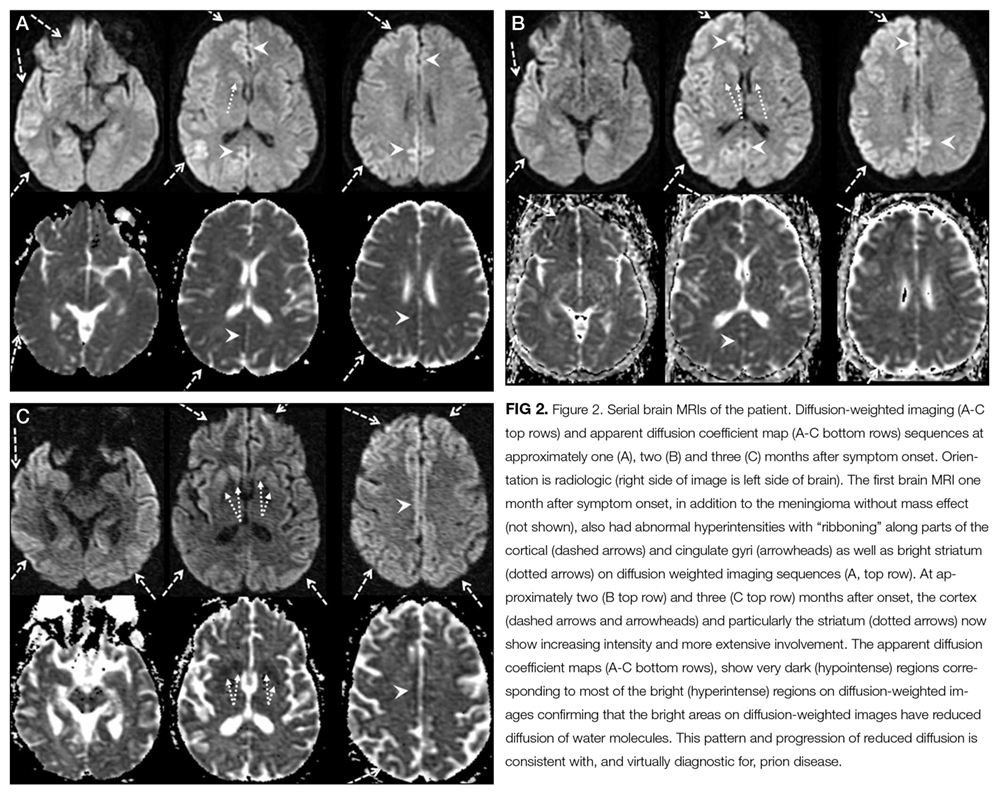
This case highlights the challenges of seeking the correct diagnosis and its consequences, especially from an infection control perspective. The initial imaging finding of a mass lesion (a meningioma—which is a common incidental finding in older adults15) was a red herring that initially obscured the correct diagnosis. The patient’s progressive cognitive decline, EEG results, and evolving MRI findings, however, prompted further scrutiny of the brain biopsy specimen that eventually steered the clinicians away from mass confusion to diagnostic certainty.
TEACHING POINTS
- Rapidly progressive dementias (RPD) are characterized by cognitive decline over weeks to months. The RPD differential diagnosis includes fulminant forms of common neurodegenerative disorders (eg, Alzheimer’s disease, dementia with Lewy bodies, frontotemporal dementia spectrum), autoimmune encephalidites, CNS cancers, and prion disease.
- Sporadic CJD is the most common human prion disease. It is a rare neurodegenerative condition with onset usually between the ages of 50 and 70 years, and most commonly manifests with rapidly progressive dementia, ataxia, and myoclonus.
- Because of its protean manifestations, the diagnosis of CJD is difficult to make antemortem, and diagnosis is often delayed. Specialist evaluation of brain MRI DWI sequences and new CSF diagnostic tests may allow for earlier diagnosis, which has management and infection control implications.