Hallmark tumor metabolism becomes a validated therapeutic target





Citation JCSO 2018;16(1):e47-e52
©2018 Frontline Medical Communications
doi https://doi.org/10.12788/jcso.0389
Submit a paper here
Altered cell metabolism has long been recognized as a distinctive feature of malignant cells but, until recently, research efforts had focused on a single aspect. It has become increasingly evident that many metabolic pathways are altered in cancer cells. Improved understanding has yielded the first regulatory approval in this new class of drugs. Here, we discuss the latest developments in the therapeutic targeting of the cancer metabolism hallmark.
A cancer cell’s sweet tooth
The metabolism of cancer cells differs from that of normal cells, an observation that has spawned a dedicated field of research and new targeted drug development. The German physiologist Otto Warburg is credited as the father of the field with his observations about the way in which cancer cells derive energy from glucose.1
In normal cells, glucose is converted into pyruvate in the cytoplasm, which is then, most often, fed to the mitochondria that use oxidative phosphorylation to produce energy in the form of adenosine triphosphate (ATP). Cancer cells seem instead to favor using the pyruvate to produce lactate through glycolysis (Figure 1).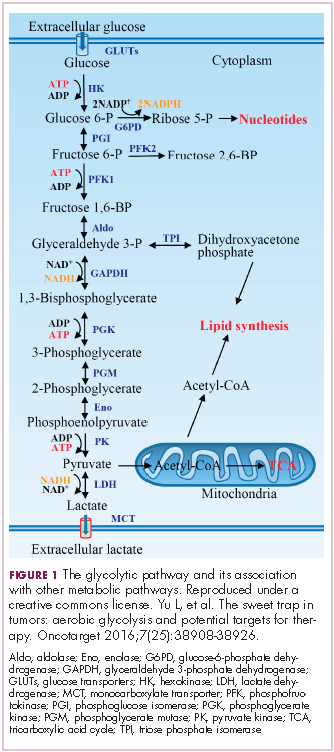
Glycolysis is usually reserved for conditions of poor oxygen availability, but although the tumor microenvironment is often hypoxic, cancer cells have been shown to use glycolysis even when oxygen is plentiful. As a result, the phenomenon is known as aerobic glycolysis, although it is most often referred to as the Warburg effect.2
Glycolysis is much less efficient than oxidative phosphorylation at producing energy, yielding only 2 ATP. In order to meet their energy demands in this way, cancer cells ramp up their glucose intake, an effect that has been exploited for the detection of cancer with positron-emission tomography.
Warburg postulated that this metabolic shift was a result of mitochondrial damage and defective oxidative phosphorylation, even going so far as to suggest that cancer was a mitochondrial disease. It has subsequently been shown that the mitochondria are mostly intact in cancer cells and that oxidative phosphorylation can still occur.3
The Warburg effect has been the subject of significant investigative efforts as researchers have attempted to better understand how this phenomenon comes about. Studies have shown that it is driven in large part by the transcription factors hypoxia inducible factor 1 alpha (HIF-1α) and c-Myc. In addition, numerous other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, and the activation of oncogenes and inactivation of tumor suppressors, are thought to play a central role.
HIF-1α is an oxygen-sensing transcription factor that coordinates cellular responses to reduced oxygen levels by binding to specific regions, known as hypoxia response elements, on target genes in the nucleus and regulating their subsequent expression. Oxygen levels and metabolism are tightly linked, and HIF-1α sits at the intersection of the 2 since many of its target genes are involved in metabolic pathways, including many glycolytic enzymes, but it also directly inhibits oxidative phosphorylation by suppressing key enzymes in this metabolic pathway.
The expression of HIF-1α and numerous glycolytic enzymes, including lactate dehydrogenase (LDH), phosphofructokinase (PFK), hexokinase II (HKII), and pyruvate dehydrogenase kinase (PDK) is increased in many tumor types. Other molecules that are associated with glucose uptake and metabolism are also dysregulated, such as the GLUT-1 glucose transporter.2,4-6
Targeting glycolysis and glucose uptake
According to one study, glucose transporters and glycolytic enzymes are overexpressed in 24 different types of cancer, representing more than 70% of all cancer cases.7 This enables cancer cells to respond metabolically as though they are experiencing hypoxia, even when oxygen is plentiful and, indeed, when hypoxia is a concern, to mount a faster response. It also provides a tempting avenue for anticancer drug design by exploiting the dependency of cancer cells on glycolysis to survive and thrive.
Inhibitors of HKII, LDH, PFK, PDK, and GLUT-1 have been and continue to be developed. For example, 2-deoxy-D-glucose is a glucose molecule in which the 2-hydroxyl group has been replaced by hydrogen, preventing further glycolysis; it acts as a competitive inhibitor of HKII. Dichloroacetate (DCA) activates the pyruvate dehydrogenase complex and inhibits the actions of the PDKs. Although development of DCA itself was unsuccessful, DCA derivatives continue to be pursued. WZB117 and STF-31 are novel small-molecule inhibitors of GLUT-1-mediated glucose transport. To date, where inhibitors of glycolysis have progressed into clinical trials, they have not proved successful, often limited by off-target effects and low potency.8-11
A variety of cell signaling pathways are implicated in metabolism by tightly regulating the ability of cells to gain access to and use nutrients. Through aberrations in these pathways, cancer cells can essentially go rogue, ignoring regulatory signals and taking up nutrients in an autonomous manner. One of the most frequently altered signaling pathways in human cancer, the PI3K-Akt-mTOR pathway, is also an important regulator of metabolism, coordinating the uptake of multiple nutrients, including glucose.
Akt in particular is thought to have a critical role in glucose metabolism and increased Akt pathway signaling has been shown to correlate with increased rates of glycolysis in cancer cells. Thus, Akt inhibitors could double as glycolytic or glucose transport inhibitors.12,13
A number of Akt inhibitors are being evaluated in clinical trials (Table) and results from the phase 2 LOTUS trial of ipatasertib (GDC-0068) were recently published.
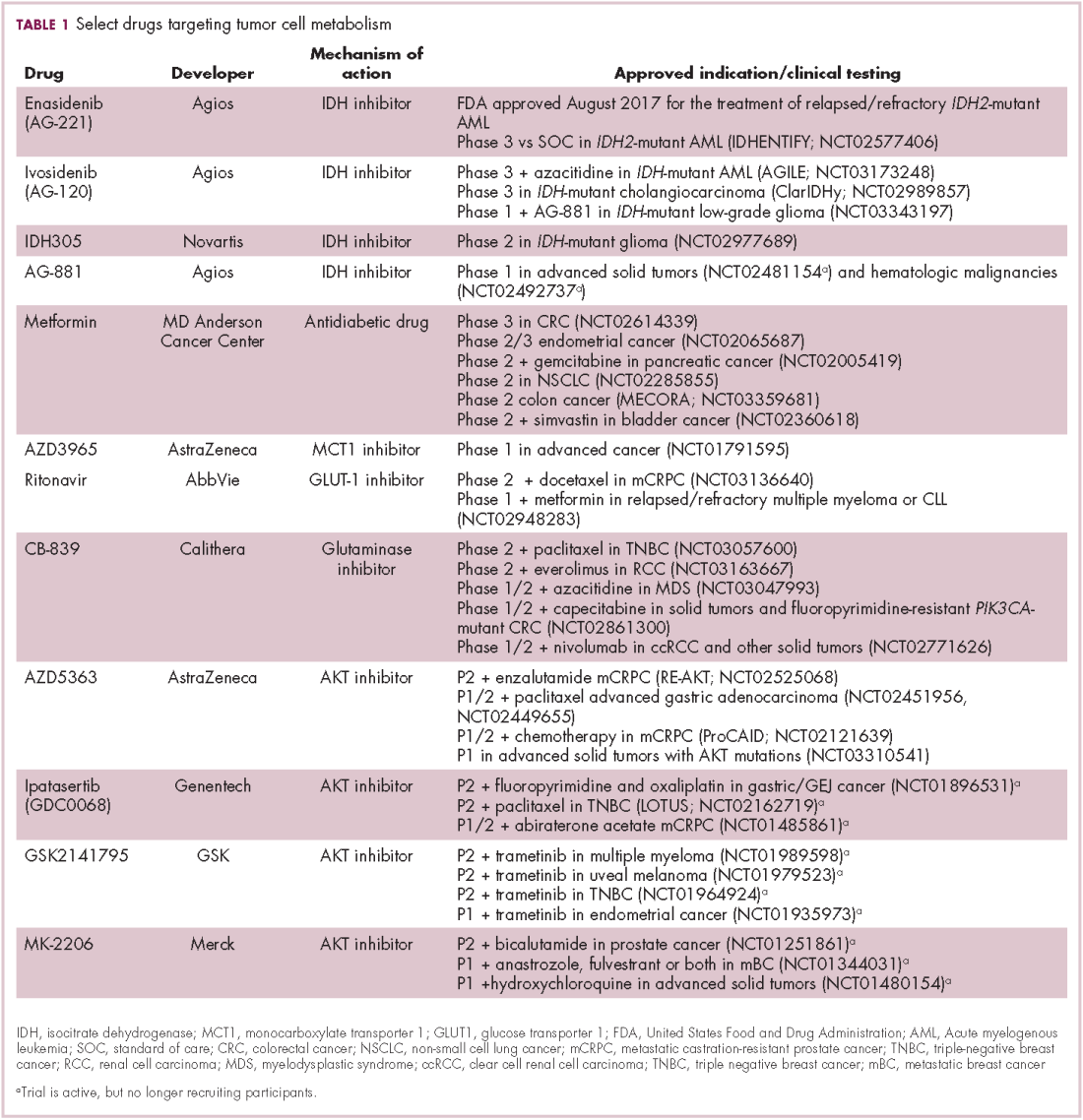
Among 124 patients randomly assigned to paclitaxel in combination with either ipatasertib or placebo, there was a modest improvement in progression-free survival (PFS) in the ipatasertib arm in patients with triple-negative breast cancer (TNBC; 6 months vs 4.2 months, respectively; hazard ratio [HR], 0.60; P = .037). The effect was more pronounced, though not statistically significant, in patients with phosphatase and tensin homolog (PTEN)-low tumors (6.2 months vs 3.7 months; HR, 0.59; P = .18). The most common grade 3 and higher adverse events (AEs) were diarrhea, reduced neutrophil count, and neutropenia.14