The Burden of Cardiac Complications in Patients with Community-Acquired Pneumonia





Pathophysiological Processes of Heart Disease Caused by CAP
The pathophysiology of cardiac complications as a result of CAP is made up of several hypotheses, including (1) declining oxygen provision by the lungs in the face of increasing demand by the heart, (2) a lack of reserve for stress because of cardiac comorbidities and (3) localized (pulmonary) inflammation leading to systemic (including cardiac) complications by the release of cytokines or other chemicals. Any of these may result in cardiac complications occurring before, during, or after a patient has been hospitalized for CAP. Antimicrobial treatment, specifically azithromycin, has also been implicated in myocardial adverse effects. Although azithromycin is most noted for causing QT prolongation, it was associated with myocardial infarction (MI) in a study of 73,690 patients with pneumonia [8]. A higher proportion of those who received azithromycin had an MI compared to those who did not (5.1% vs 4.4%; OR 1.17; 95% CI, 1.08–1.25), but there was no statistical difference in cardiac arrhythmias, and the 90-day mortality was actually better in the azithromycin group (17.4% vs 22.3%; odds ratio [OR], 0.73; 95% CI, 0.70–0.76).
Systemic inflammation is the result of several molecules, such as cytokines, chemokines and reactive oxidant species. Reactive oxidant species may determine oxidation of proteins, lipids and DNA, which leads to cell death. The hypothesis also purports that they also cause destabilization of atherosclerotic plaques leading to MIs. Other reactions as a result of inflammation lead to arrhythmias with or without compromised cardiac function, causing congestive heart failure (CHF). For this reason, some authors have approached the pathophysiology of cardiac complications by considering them to be either plaque-related or plaque-unrelated events [9].
A few studies have linked specific inflammatory molecules to cardiac toxicity. NOX2 is chemically unstable and may provoke cellular damage, thus maintaining a certain redox balance is crucial for cardiomyocyte health. In 248 patients with CAP, an elevated troponin T was present in 135 patients and among those, NOX2 correlated with the troponin T values (OR 1.13, 95% CI 1.08–1.17; P < 0.001) [10]. Both disrupting the equilibrium of the redox balance by upregulating NOX2, and finding NOX2 to be associated with troponin T suggest that oxidative stress is implicated in damage to the myocardium during CAP. In another study of 432 patients with CAP, 41 developed atrial fibrillation within 24 to 72 hours of admission and showed higher blood levels of NOX2 than those who had CAP without atrial fibrillation [11]. Oxidative stress has been shown to cause hypertrophy, dysfunction, apoptotic cell death, and fibrosis in the myocardium [12].
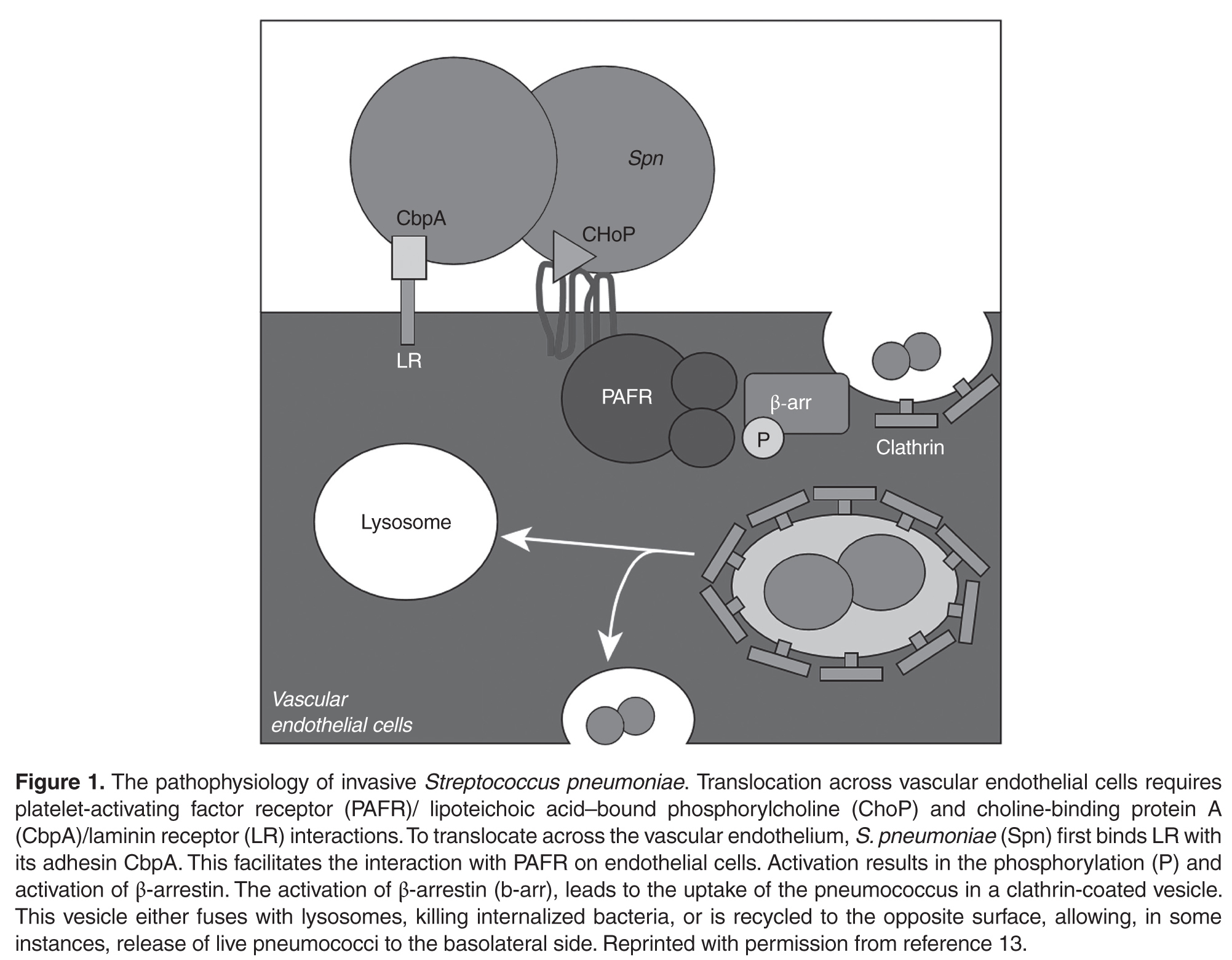
There is likely a high level of variability in how individual patients respond to a predisposing factor for a cardiac complication. For example, one patient may tolerate a mild hypoxia while another is sensitive. The association of inflammatory markers with the presence of cardiac markers, however, would support that once there are systemic reactions, the complications increase. Macrolides, however, were not found to contribute to long-term mortality due to cardiac complications.
Cardiac Complications of CAP
After the H1N1 influenza outbreak of 1918, it was noted that all-cause mortality increased during the outbreak as did influenza-related deaths. This prompted inquiry as to whether there was an actual association between the outbreak and increased overall mortality, or whether the 2 occurrences were simply coincidental [14]. Near that time, arrhythmias in CAP patients were studied. T-wave changes were found to be associated with CAP [15]. Among 92 patients studied, 449 electrocardiograms (ECGs) were reviewed. T-wave changes were the most common ECG changes. They were found in 5 of 10 of the patients who died, and in 35 of the 82 patients who lived. Twelve living patients had persistent ECG changes, and although they were all thought to have had underlying myocardial disease, 2 of them certainly did as they each had an acute MI (and the ECG was included as a figure for one of them).
A study in the 1980s that reported 3 of 38 CAP patients with CHF interrupted the paucity of data at the time that showed that having a cardiac complication during CAP was a known entity [16]. By the end of the 20th century, Meier et al noted that among case patients who had an MI, an acute respiratory tract infection preceded the MI in 2.8% while in only 0.9% of control patients [17]. They also noted that patients who had an acute respiratory tract infection were 2.7 times more likely to have an MI in the following 10 days than control patients.
 Further study by Musher et al revealed that MI was associated with pneumococcal pneumonia in 12 (7%) of 170 veteran patients [18]. An MI was defined on the basis of ECG abnormalities (Q waves or ST segment elevation or depression) with troponin I levels ≥ 0.5 ng/mL. They also evaluated arrhythmias and CHF. They included atrial fibrillation or flutter and ventricular tachycardia while excluding terminal arrhythmias. An arrhythmia was found in 8 (5%) patients. CHF was based on Framingham criteria (Table 1) [19]. New or worsening CHF was determined by comparing physical findings, laboratory values, chest radiograph, and echocardiogram reports in medical records. CHF was found in 13 (19%) patients. Ramirez et al found that MI was associated with CAP in 29 (5.8%) of 500 similar veteran patients [20].
Further study by Musher et al revealed that MI was associated with pneumococcal pneumonia in 12 (7%) of 170 veteran patients [18]. An MI was defined on the basis of ECG abnormalities (Q waves or ST segment elevation or depression) with troponin I levels ≥ 0.5 ng/mL. They also evaluated arrhythmias and CHF. They included atrial fibrillation or flutter and ventricular tachycardia while excluding terminal arrhythmias. An arrhythmia was found in 8 (5%) patients. CHF was based on Framingham criteria (Table 1) [19]. New or worsening CHF was determined by comparing physical findings, laboratory values, chest radiograph, and echocardiogram reports in medical records. CHF was found in 13 (19%) patients. Ramirez et al found that MI was associated with CAP in 29 (5.8%) of 500 similar veteran patients [20].
Corrales-Medina et al reported cardiac complications in CAP patients in the Pneumonia Patient Outcomes Team cohort study [21]. They defined MI as the presence of 2 of 3 criteria: ECG abnormalities, elevated cardiac enzymes, and chest pain. They found 43 (3.2%) of 1343 patients with an MI. Arrhythmias included atrial fibrillation or flutter, multifocal atrial tachycardia, supraventricular tachycardia, ventricular tachycardia (≥ 3 beat run) or ventricular fibrillation. With the more inclusive list, they found a greater proportion, 137 (10%) patients affected. They defined CHF with physical examination findings plus a radiographic abnormality, and found 279 (21%) patients affected. A meta-analysis of 17 studies had pooled incidences for an MI of 5.3%, an arrhythmia of 4.7% and CHF of 14.1% [22].
In summary, the most prominent cardiac complications in patients with CAP have been found to be CHF, MI, and arrhythmia.
Timing of Cardiac Complications in Relation to CAP
While a patient is still in the community, cardiac complications may occur with the onset of CAP, or afterwards. For these patients, the primary goal is to identify the complication and manage it as soon as the patient is admitted for CAP, rather than allowing the complications to worsen only to be recognized later. Cardiac complications are rare in outpatients overall. A study of 944 outpatients found heart failure in 1.4%, arrhythmias in 1.0% and MI in 0.1% [21].
For patients who are admitted with CAP but who do not have a cardiac complication, the goals are either to prevent any complication or to recognize and manage a complication early. This also applies to patients who have been discharged after an admission for CAP. Cardiac complications have been recorded shortly after (within 30 days), and late (up to 1 year) after discharge. A study of over 50,000 veterans who were admitted for CAP were followed for any cardiovascular complication in the next 90 days. Approximately 7500 veterans were found to have a cardiac complication, including (in order of highest to lowest frequency) CHF, arrhythmia, MI, stroke and angina [23]. More than 75% of the complications were found on the day of hospitalization, but events were still measured at 30 days and 90 days.
Two other studies sought to determine an association between CAP and cardiac complications differently; not by following CAP patients prospectively for complications but by retrospectively evaluating patients for a respiratory infection among those who were admitted for a cardiovascular complication (MI or stroke). A study of over 35,000 first-time admissions for either an MI or a stroke were evaluated for a respiratory infection within the previous 90 days [24]. The incidence rates were statistically significant for every time period up to 90 days. The preceding 3 days was the time period with the highest frequency for a respiratory infection preceding an event. When the event was an MI, the incident rate was 4.95 (95% CI, 4.43–5.53). A similar study of over 20,000 first-time admissions for either an MI or stroke were evaluated for a preceding primary care visit for a respiratory infection [25]. An infection preceded 2.9% of patients with an MI and 2.8% of patients with a stroke. Statistical significance was found for the group of patients who had a respiratory infection within 7 days preceding an MI (OR 2.10 [95% CI 1.38–3.21]) or preceding a stroke (OR 1.92 [95% CI 1.24–2.97]). In fact, every time period analyzed for both complications (MI and stroke) was significant up to 1 year. Because the timing of a cardiac complication varies and can occur up to 90 days or even a year after acute infection, physicians should maintain vigilance in suspecting and screening for them.
Predictors of Cardiac Complications During CAP
Recently, Cangemi et al reviewed mortality in 301 patients admitted for CAP 6 to 60 months after they were discharged [26]. Mortality was compared between patients who experienced a cardiac complication—atrial fibrillation or an ST- or non-ST-elevation MI—during their admission and those who did not. A total of 55 (18%) patients had a cardiac complication while hospitalized. During the follow-up, 90 (30%) of the 301 patients died. Death occurred in more patients who had had a cardiac complication while hospitalized than in those who did not (32% vs 13%; P < 0.001). The study also showed that age and the pneumonia severity index (PSI) predicted death in addition to intra-hospital complication. A Cox regression analysis showed that intrahospital cardiac complications (hazard ratio [HR] 1.76 [95% CI 1.10–2.82]; P = 0.019), age (HR 1.05 [95% CI 1.03–1.08]; P < 0.001) and the PSI (HR 1.01 [95% CI 1.00–1.02] P = 0.012) independently predicted death after adjusting for possible confounders [26].
The PSI score was published in 1997, and it instructed that patients with a risk class of I or II (low risk) should be managed as outpatients. Data eventually showed that there is a portion of the population with a risk class of I or II whose hospital admission is justified [4]. Among the reasons found was “comorbidity,” including MI and other cardiac complications. The PSI prediction rule was found to be useful in novel ways, and being associated with a risk of MI in patients with CAP was one of them. The propensity-adjusted association between the PSI score and MI was significant (P < 0.05) in an observational study of the CAP Organization (CAPO) [20]. Knowing that a PSI of 80 is in the middle of risk class III (71–90), it was noted that below 80 the risk for MI was zero to 2.5%, while above 80 the risk rose from 2.5% to 12.5%. A later study using the same statistical method showed a correlation between the PSI score and cardiac complications (MI, arrhythmias and CHF) with a P value of < 0.01 [21]. Determining the probability for the combination of complications, rather than just an MI, yielded an unsurprisingly higher range of risk for the PSI below 80, which was zero to 17.5%, while risk for a PSI above 80 was 17.5% to 80%.
In a study to determine risk factors for cardiac complications among 3068 patients with CAP, Griffin et al applied a purposeful selection algorithm to a list of factors with reasonable potential to be associated with the 376 patients who actually had a cardiac complication [27]. After multivariate logistic regression analysis, hyperlipidemia, an infection with Staphlococcus aureus or Klebsiella pneumoniae, and the PSI were found to be statistically significant. In contrast, statin therapy was associated with a lower risk of an event.
In 2014, a validated score similar to the PSI and using the same database was derived to predict short-term risk for cardiac events in hospitalized patients with CAP [28]. It attributes points for age, 3 preexisting conditions, 2 vital signs and 7 radiological and laboratory values, with a point scoring system that defines 4 risk stratification classes. In the derivation cohort, the incidence of cardiac complications across the risk classes increased linearly (3%, 18%, 35%, and 72%, respectively). The score was validated in the original publication with a separate database but has not been evaluated since. The score outperformed the PSI score in predicting cardiac complications in the validation cohort (proportion of patients correctly reclassified by the new score, 44%). Potentially, the rule could help identify high-risk patients upon admission and could assist clinicians in their decision making.