Management of Relapsed and Refractory Multiple Myeloma





Case Studies
Patient A
A 62-year-old man with IgG-kappa MM was diagnosed 4 years ago during evaluation of a pathologic humeral fracture. The disease was prognostically standard risk, with revised International Staging System (RISS) stage I disease (beta-2 microglobulin 3.4 mcg/mL, albumin 4.1 g/dL, normal cytogenetics with 46,XY in 20 cells analyzed, and myeloma fluorescent in situ hybridization [FISH] panel showing t(11;14) but no del17p, t(14;16), t(14;20), or t(4;14)) [5], and normal blood counts, organ function, and lactate dehydrogenase (LDH) at diagnosis. He was treated with 5 cycles of standard lenalidomide, bortezomib, and dexamethasone followed by high-dose melphalan with autologous stem cell transplantation (ASCT) and then lenalidomide continuous maintenance. He achieved a stringent complete response (ie, complete disappearance of myeloma-derived monoclonal proteins in the serum and urine, a normal serum free light chain ratio, and undetectable monoclonal plasma cells on a bone marrow aspirate and biopsy) [4]. His MM was monitored every 2 to 3 months for disease progression and medication toxicity. At month 38, a monoclonal protein spike (M-spike) on serum protein electrophoresis (SPEP) remained undetectable, but serum kappa free light chain levels increased from 1.98 mg/dL to 8 mg/dL with stable lambda serum free light chains and a ratio that rose to 16, consistent with low-level biochemical recurrence. He had no evidence of end-organ damage and therefore was maintained on lenalidomide maintenance for the time being. Over the next 12 months, his kappa serum free light chain level continued to slowly rise, reaching 24 mg/dL, while the ratio rose to 50. There was still no detectable M-spike. He developed mild anemia during this time, with his hemoglobin dropping from a prior value of approximately 11 g/dL to 9.8 g/dL, though kidney function remained normal. A repeat bone marrow aspirate and biopsy revealed 20% kappa-restricted plasma cells.
Patient B
A 75-year-old woman with IgA-kappa MM was diagnosed after laboratory testing by her primary care physician incidentally showed an elevated serum total protein level. The MM was intermediate risk, with RISS stage II disease, and with mild renal impairment resulting in an estimated creatinine clearance of 45 mL/min that was felt to be due to MM. She was initially treated with bortezomib and dexamethasone but received only 2 cycles because she developed painful peripheral neuropathy secondary to bortezomib. Bortezomib was stopped and she was then treated with lenalidomide and dexamethasone for 4 cycles. She achieved a complete response and elected to stop treatment due to fatigue. Her fatigue did not improve off treatment. Six months after stopping therapy, an M-spike was detectable at 0.1 g/dL and she developed a new painful lytic lesion in the left humerus.
Patient C
A 59-year-old man with lambda free light chain MM was diagnosed when he presented with acute renal failure requiring dialysis. The disease was RISS-III at diagnosis (high risk), with the t(4;14) genetic abnormality in his MM cells detected on bone marrow aspirate, an abnormality that has been associated with poor prognosis MM [6–8]. The patient was treated with cyclophosphamide, bortezomib, and dexamethasone [9] for 6 cycles, at which point his disease was in a very good partial response (>90% reduction in M-spike) [4], and his renal function had recovered to a new baseline creatinine clearance of 45 mL/min. He then underwent ASCT after melphalan conditioning followed by bortezomib maintenance therapy every 2 weeks. Eight months after ASCT, his lambda free light chain level increased from 1.25 mg/dL to 45 mg/dL and the ratio increased from 4 to 22. Renal function was unchanged and there was stable anemia, with hemoglobin of 10.1 g/dL.
When should treatment for RRMM commence?
Patients with MM in remission are closely monitored, with clinical and laboratory examinations generally conducted every 1 to 3 months. The history is focused on MM-related symptoms such as increasing bone pain or weight loss, and symptoms of therapy-related toxicity such as fatigue, gastrointestinal distress, or peripheral neuropathy. Laboratory assessment typically includes blood counts and chemistry measurements, as well as measurements of MM-derived monoclonal proteins: SPEP, serum immunofixation (IFE), serum immunoglobulin free light chain measurements, and urine protein electrophoresis and immunofixation (UPEP/urine IFE) [10]. Progressive disease biochemically is defined as a 25% increase in M-spike (at least 0.5 g/dL if the M-spike is in serum or > 200 mg/24 hours if in urine), and/or a rise of greater than 10 mg/dL difference between the involved and uninvolved serum free light chains. Clinically progressive disease is denoted as new evidence of end-organ damage such as a new plasma-cytoma, unexplained hypercalcemia, or worsening anemia due to MM [4]. Many, if not most, patients will have biochemical recurrence identified by laboratory measurements ofmonoclonal proteins before clinical recurrence transpires.
The velocity of relapse can help guide decisions about when to reinitiate therapy. High-velocity disease relapse, meaning rapid rise in monoclonal proteins, is an indicator of more aggressive disease, and treatment should be initiated promptly before development of symptoms [11]. Conversely, low-level, indolent recurrence can often be followed with a “watch and wait” approach to determine how the myeloma will progress over time. Expert guidelines suggest that a monoclonal protein doubling time of 2 months may be an appropriate cutoff for determining high versus low velocity [12], although 2 months is not a firm rule and the decision of when to restart treatment for any given patient with asymptomatic biochemical recurrence should be individualized. Importantly, it is not clear that changing therapy at the time of biochemical recurrence, prior to clinical disease progression, improves outcomes, but clinicians are often nonetheless hesitant to hold therapy in the face of biochemically recurrent MM given the potential for complications, such as a pathologic fracture. In patients with biochemically recurrent MM for whom re-initiation of systemic anti-myeloma therapy is being deferred, one can consider re-initiation of zoledronic acid therapy, since in a randomized controlled trial, zoledronic acid commenced at the time of biochemical relapse resulted in fewer skeletal events as compared to placebo [13].
What disease factors should be considered in choosing treatment for RRMM?
MM exhibits genetic complexity, and prior treatments may result in clonal evolution of and selection for an initially nondominant, treatment-resistant clone [14,15]. This heterogeneity and selection pressure may explain why 3-drug regimens often outperform 1- or 2-drug regimens, why each remission is generally shorter than the last, and why patients who have enjoyed a long duration of response to one therapy and been off it for some length of time may again have a good response when re-treated with the same therapy at time of MM relapse. So how does one know if a new clone has emerged? While there is no standard for monitoring intra-clonal heterogeneity presently, changes in clinical phenotype likely correlate with evolving clones. Some such changes include free light chain escape (ie, MM that initially secreted an intact M-spike and then only secretes free light chain at relapse), new development of extramedullary disease (plasmacytomas outside of bone) in patients who previously had MM only in the bone marrow, and resistance of some sites to treatment while others respond (a mixed response). The former 2 phenotypes in particular portend poor prognosis and unsurprisingly they can be seen together [16–19]. Restaging, meaning a complete reassessment of MM disease status at the time of relapse, including bone marrow aspirate and biopsy, is beneficial to help guide therapy, as those with high-risk features including high ISS stage [20], high-risk cytogenetics, increased LDH, and extramedullary disease should be treated with triplet therapy when possible [11]. Repeat imaging should also be considered as a new baseline comparator. This can be done with standard x-rays, positron-emission tomography/computed tomography (PET-CT), or magnetic resonance imaging. PET-CT offers the advantage of showing active disease sites and the presence of extramedullary disease, although it exposes the patient to more radiation than the other methods.
In terms of using genetics to guide therapy decisions in RRMM, the presence of the del(17p) abnormality either by karyotyping or FISH portends high risk and pomalidomide in one study was shown to mitigate that risk [21]. How genetics and prognostic markers should dictate therapy selection in RRMM otherwise, however, is unclear and an area of active research efforts.
What patient factors should be considered in choosing treatment?
Given the relatively large selection of possible regimens for the treatment of RRMM, patient preference can be incorporated into regimen selection. Patients who have long commutes or who are trying to work may not be ideal candidates to receive carfilzomib-based regimens given the twice-per-week infusion schedule (though a once-a-week dosing schedule is being tested) [22]. Patients who have poor venous access may be good candidates for all-oral regimens. Prior treatment tolerability and side effects should also be considered. Patients who experienced significant peripheral neuropathy with bortezomib may have less neuropathy with carfilzomib. Those with renal failure may tolerate pomalidomide better than lenalidomide [23].
Patient age and functional status are important considerations in choosing a treatment regimen for RRMM. Very old patients (a subjective categorization to include patients > 80 years by chronologic or physiologic age), those with functional dependence, or patients harboring substantial medical comorbidities are at risk for therapy toxicity and so often warrant less intensive approaches [24]. Deciding which patients empirically warrant less dose-intensive approaches can be challenging, especially with the growing recognition that fit seniors can often tolerate and enjoy the benefits of full-dose approaches, including sometimes even ASCT. Geriatric assessment instruments that interrogate a variety of geriatric-relevant domains, such as number of falls, independence in activities of daily living, and polypharmacy, are being investigated as toxicity predictors and may help make those decisions in the future. Such instruments have been shown to predict chemotherapy toxicity in solid tumors [25,26] and preliminarily in MM [27], but they remain investigational. While no validated geriatric assessment instruments are currently available for routine clinical employment in MM, clinicians should consider the geriatric domains that these instruments assess when choosing among treatment options. Clinically, that often translates to choosing gentler regimens with likely better tolerability, albeit perhaps with less efficacy, for patients judged to be vulnerable to toxicity.
As part of therapy selection in RRMM, the clinician needs to consider if the patient is a candidate for ASCT. For patients who did not undergo ASCT as part of initial treatment, ASCT can be considered at the time of relapse. Ideally, all patients who could eventually undergo ASCT should have hematopoietic stem cells collected and stored at the time of first induction; however, collection after re-induction chemotherapy has been shown to be feasible [28,29]. ASCT for RRMM appears to be effective, although rigorous randomized comparisons of ASCT versus treatment purely with novel drugs are lacking [30–32]. For patients who did receive ASCT consolidation in the frontline, if a response is sustained for 18 months or greater, existing guidelines suggest that a second ASCT is likely worthwhile [29]. Whether the routine usage of maintenance therapies (low-dose, usually single drugs used to prolong duration of remission once remission is achieved) should change that 18-month cutoff is unclear, however, since maintenance “artificially” makes ASCT appear more effective by prolonging post-ASCT duration of remission. The “is it worth it” discussion is also largely subjective and hinges heavily on the patient’s experience with the first ASCT. In our practice, we often use 3 years as the cutoff for considering repeat ASCT in patients on maintenance therapy, meaning that if a patient underwent ASCT and received maintenance, a remission lasting more than 3 years means we consider ASCT as part of therapy for relapse.
Allogeneic stem cell transplantation (allo-SCT) is a treatment option for RRMM generally reserved for fit patients younger than 65 years [22,33]. The timing of allo-SCT is also controversial, with some reserving it as a last option given a historically high transplant-related mortality and improved progression-free survival but not necessarily overall survival benefit. A recent consensus statement has suggested allo-SCT be considered (preferentially in a clinical trial) for eligible patients with high-risk disease who relapse after primary treatment that included ASCT [29]. With the abundance of new treatment options in RRMM with reasonable toxicity profiles, it is not clear for whom and when allo-SCT is best considered.

Which regimen should be used to treat a first relapse?
Entry into a well-designed clinical trial for patients with RRMM should be considered for every patient since there is a lack of evidence to guide the best sequencing of chemotherapies [11]. Beyond that, the choice of therapy is based upon 2 main factors: the disease itself (eg, indolent, asymptomatic biochemical recurrence versus aggressive clinical recurrence with new fractures or extramedullary plasmacytomas), and the patient’s preferences and characteristics, such as age, performance status, comorbidities, and toxicities from prior therapies. In looking for the “best” re-induction regimen, it is tempting to compare the efficacy of regimens across trials, but such efforts are fraught given the significant heterogeneity of the patient populations between trials. As an example, comparing daratumumab + pomalidomide + dexamethasone (DPd) to daratumumab + lenalidomide + dexamethasone (DRd), one may conclude that DRd is superior, given an overall response rate of 88% in DRd versus 58% in DPd. However, the DPd trial included patients who were refractory to lenalidomide and bortezomib, while the DRd study required only treatment with one prior therapy [34,35].
For patients who enjoyed a long remission after any particular chemotherapy regimen with good tolerability and with indolent features at the time of relapse, re-treating with the same regimen can be considered, although nowadays with so many new and highly potent agents available such “backtracking” is less common and some studies suggest that employing new agents may be beneficial. As an example, in the randomized ENDEAVOR study of bortezomib + dexamethasone versus carfilzomib + dexamethasone in RRMM, 54% of patients had been exposed to bortezomib whereas virtually none had received carfilzomib prior to study enroll-ment. Among those patients with prior bortezomib exposure, median progression-free survival was 15.6 versus 8.1 months (hazard ratio 0.56, [95% confidence interval 0.44 to 0.73]) for carfilzomib versus bortezomib, respectively. Follow-up was too immature for definitive conclusions to be drawn about overall survival, but the substantial difference in progression-free survival provides a compelling argument for using carfilzomib instead of going back to bortezomib for patients with prior bortezomib exposure [36].
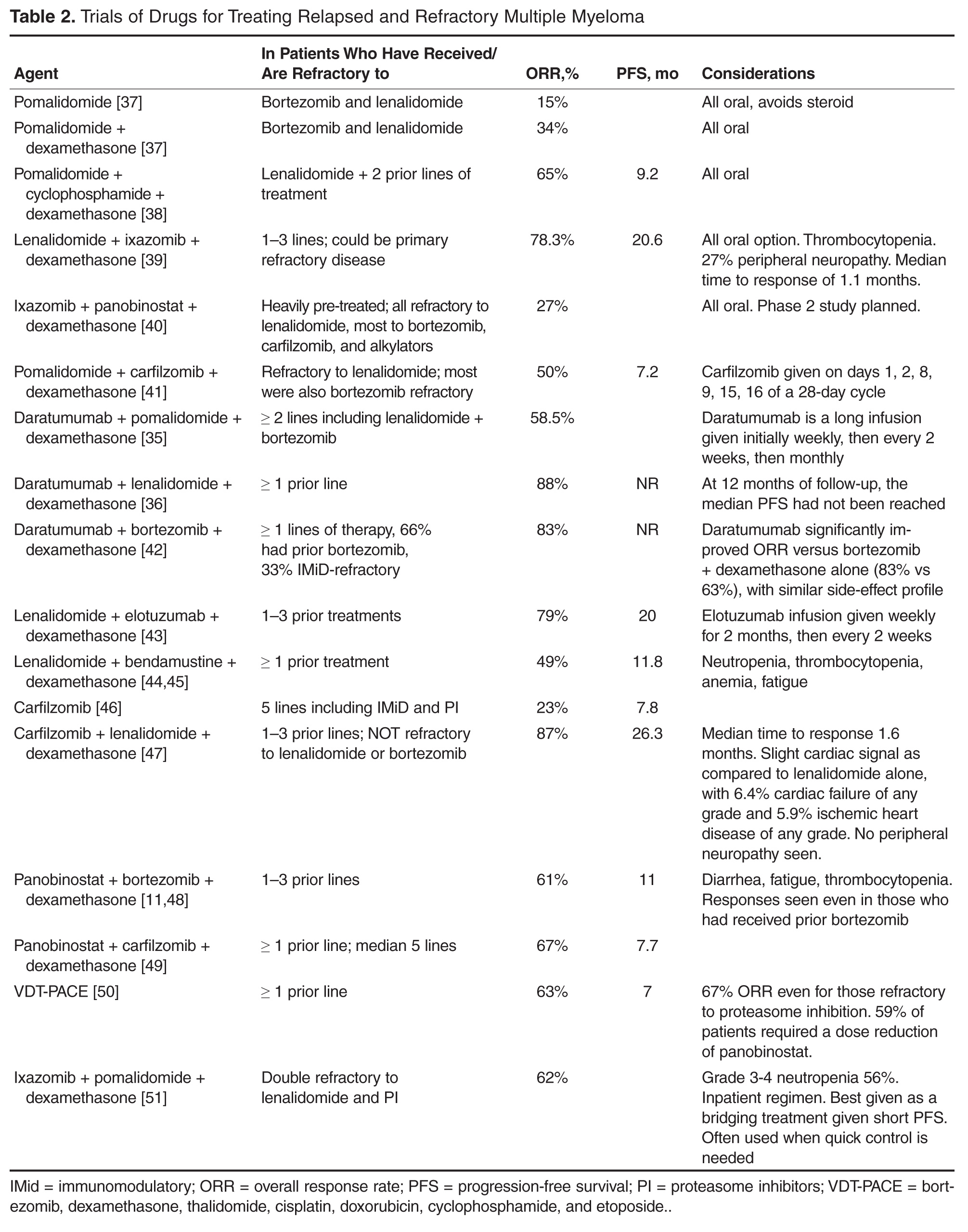
For patients who are fit and not very old, we generally employ triplet re-induction. For the large number of these patients who were previously exposed to both lenalidomide and bortezomib, including as part of a maintenance strategy, outside of clinical trials we routinely use carfilzomib + pomalidomide + dexamethasone [41]. For patients who are lenalidomide-naïve but bortezomib-exposed, we often employ carfilzomib + lenalidomide + dexamethasone based on the phase 3 ASPIRE trial, which showed a significantly improved progression-free survival with carfilzomib + lenalidomide + dexamethasone versus lenalidomide + dexamethasone [47]. For patients who have previously received lenalidomide but not bortezomib, we consider pomalidomide + bortezomib + dexamethasone [52]. These regimens take advantage of the arguably most potent, most proven drugs in treating RRMM, namely proteasome inhibitors (bortezomib and carfilzomib) and immunomodulatory agents (lenalidomide and pomalidomide).
For patients who are more vulnerable to toxicity due to advanced age or comorbidities, we consider less intensive regimens, including dose-reduced triplets or doublets. Patients who had received lenalidomide-based combinations but not bortezomib are considered for a bortezomib-based re-induction, including bortezomib + dexamethasone alone. In the case of someone who had initially received a bortezomib-based combination but no lenalidomide, the new drugs are viable options: ixazomib [53] or elotuzumab [43] can both be added to standard lenalidomide + dexamethasone, with expectations of increasing response rates and progression-free survival and an acceptably low increased risk of severe toxicity. Ixazomib + lenalidomide + dexamethasone also has the benefit of being all-oral. For patients with bortezomib- and lenalidomide-exposed RRMM, using carfilzomib [54] or pomalidomide [55] with dexamethasone is reasonable.
Once MM has progressed beyond the arguable “core drugs” of early-stage MM, namely lenalidomide, bortezomib, carfilzomib, and pomalidomide, off protocol we favor daratumumab monotherapy [56,57]. Other options include panobinostat (given usually with bortezomib) [58] and bendamustine [59], among others.