Febrile Seizures: Evaluation and Treatment





What treatment options are available?
Complications with prolonged and/or recurrent seizures can occur. Treatments options can be stratified into 3 possible categories: emergency rescue treatment for prolonged or a cluster of febrile seizures, intermittent treatment at the time of illness, and chronic use of medication. Treatment options for complex febrile seizures may include the use of a rescue seizure medication when the febrile seizure is prolonged. Rectal preparations of diazepam gel can be effective in stopping an ongoing seizure and can be provided for home use in patients with known recurrence of febrile status epilepticus [3]. For children and adolescents where a rectal administration is not ideal, intranasal versed can be utilized instead of rectal diazepam. In addition, the use of an intermittent benzodiazepine at the onset of febrile illness can also be considered a treatment option. Using oral diazepam at the time of a febrile illness has been demonstrated in reducing the recurrence of febrile seizures [3]. Other studies have shown similar results when using buccal midazolam [32]. No adequate studies have been performed using second- or third-generation anti-epilepsy medications in the treatment of recurrent of complex febrile seizures [3].
It is unclear whether benefit is present to using intermittent benzodiazepine doses prior or during a febrile illness for those prone for recurrent febrile seizures [33]. Physicians may consider this option in patients with frequent recurrent seizures, when caregivers can identify the fever before the seizure occurs.
Overall, parental education of efficacy and side effect profiles should be discussed in detail when considering any treatment options for complex febrile seizures [34]. It is important to remember that the long-term prognosis in terms of developing epilepsy or neurological and cognitive problems is not influenced by the use of antiepileptic medications for recurrent febrile seizures [17]. Even in the case of prolonged febrile seizures in otherwise neurodevelopmentally normal children, antiepileptics have not been shown to cause damage to the brain [19].
Febrile Status Epilepticus
Febrile status epilepticus is a subtype of complex febrile seizures and is defined as a febrile seizure lasting greater than 30 minutes. Overall, febrile status epilepticus accounts for approximately 5% of all presentations of febrile seizures [35]. It represents about 25% of all episodes of childhood status epilepticus and more than two-thirds of cases during the first 2 years of life. Literature suggests that an increased risk for focal epilepsy exists [36]. Children presenting with febrile status epilepticus are more likely to have a family history of epilepsy and a history of a previous neurological abnormality [22]. It is likely to reoccur if the first presentation was febrile status epilepticus. However, increased risk for death or developmental disability as a result of the seizure is not seen [37].
The prospective multicenter study of the consequences of prolonged febrile seizures in childhood (FEBSTAT) has been conducted. The study reported that febrile status epilepticus is usually focal (67% of episodes), occurs in very young children (median age 1.3 years), and is frequently the first febrile seizure [22]. In this study, the median duration of the seizure was about 68 minutes and 24% of children had an episode lasting more than 2 hours. In 87% of the events, seizures did not stop spontaneously and benzodiazepines were needed. Focal features observed were eye and head deviation, staring, and impaired consciousness prior to the seizure and an asymmetric convulsion or Todd’s paresis.
Case 3: Epilepsy Syndromes Associated With Febrile Seizures
A 1-year-old female presents for evaluation of seizures that began at age 8 months. Seizures are described as occurring in the setting of fever with bilateral symmetric tonic clonic activity lasting durations of less than 10 minutes on average, but at least 2 instances of seizure lasting 20 minutes or more. The family notes that seizures have occurred almost every time the child has had a febrile illness and often cluster over several days. They report at least 1 seizure that occurred in the absence of fever. Development has been normal to date and an EEG done by their primary provider was also normal.
What epilepsy syndromes are associated with febrile seizures?
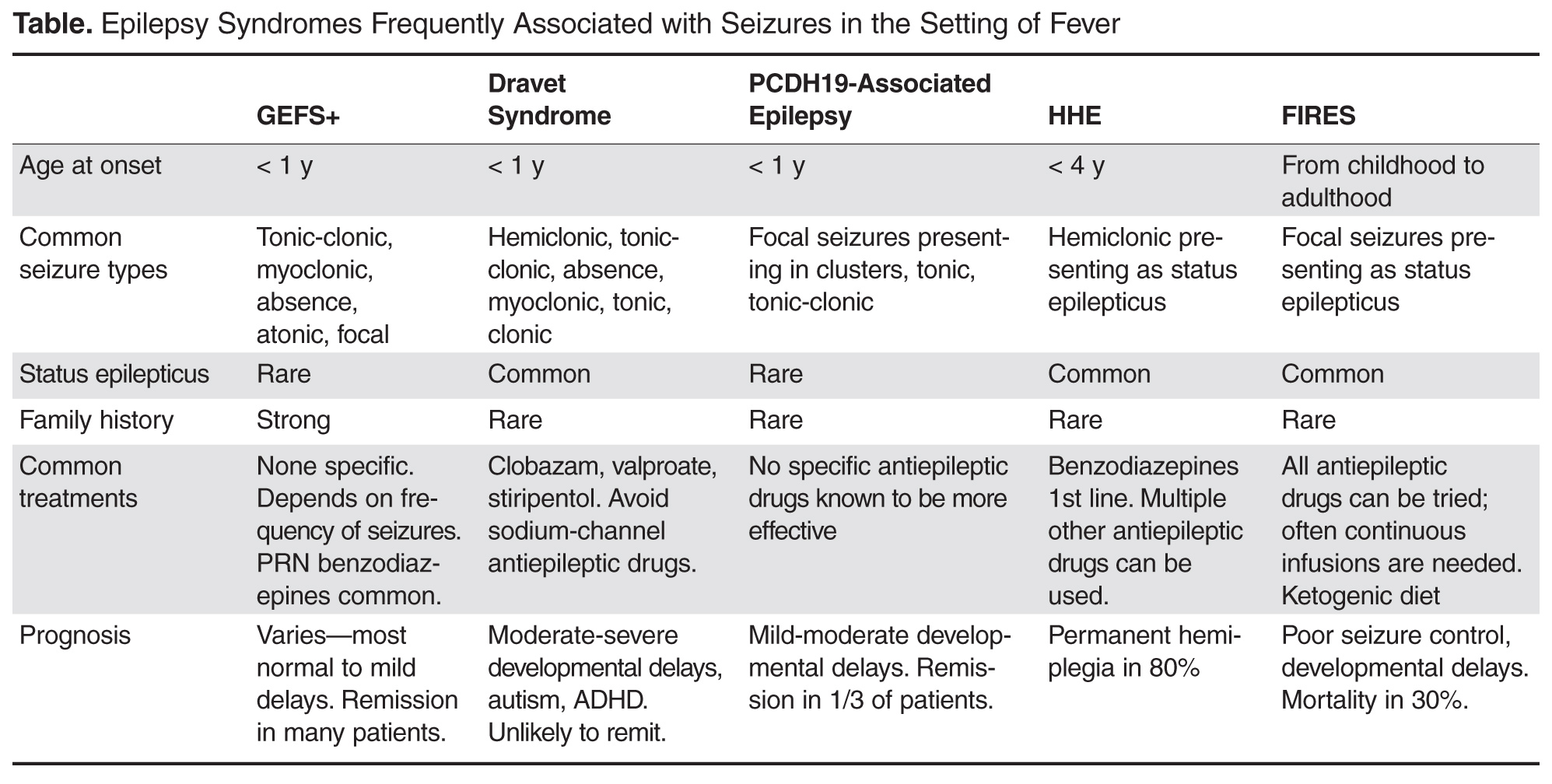
Genetic Epilepsy with Febrile Seizures Plus
GEFS+ was first described in 1997 following recognition of a pattern of febrile seizures followed later by the development of various epilepsy syndromes within the same family [38]. As such, the syndrome is defined based on the familial occurrence of febrile and afebrile seizures in at least 2 family members and can have a wide range of phenotypes. The most common presentation is of typical febrile seizures which can persist beyond the typical upper age limit of 6 years. Unprovoked generalized seizures of multiple types (ie, myoclonic, absence, atonic) occur at a later age, though focal seizures may also be present. The presence of focal onset seizures led to the naming change from “generalized” epilepsy with febrile seizures plus as it was previously referred. Seizure frequency and severity may vary between family members, as can response to treatment, making prognosis difficult to predict. As even in typical febrile seizures a family history of febrile seizure may be common, it may be difficult to diagnose the syndrome after the initial febrile seizure. However, if the family history is strong for a family member with a GEFS+ phenotype, one can appropriately counsel the family on the possibility that a similar course may evolve. While the majority of GEFS+ patients have milder phenotypes, some more severe phenotypes can have cognitive delays. Dravet syndrome falls within the spectrum of GEFS+ and is a prime example of the phenotypic continuum to more severe presentations in some patients.
The syndrome is believed to be inherited in an auto-somal dominant fashion with incomplete penetrance. Multiple genes have been implicated as a cause, though only 11.5% of families with clinical GEFS+ may have mutations [39]. SCN1A, encoding the α-subunit of the voltage-gated sodium channel is most frequently reported in GEFS+ families, yet is only found in 10% [38]. When associated with GEFS+, SCN1A mutations are more often missense type, whereas truncating and nonsense mutations are more commonly encountered in Dravet syndrome. Mutations in SCN1B encoding the β1 subunit of the voltage-gated sodium channel has also been reported [40]. Finally, the GABA(A) receptor gamma 2 subunit GABRG2 has been found in < 1% of GEFS+ families [39]. The variability in causative genes underscores the reasons for phenotype variability and it is likely that other modifier genes are responsible for the heterogeneity within GEFS+ families [41].
Dravet Syndrome
Dravet syndrome, often referred to as severe myoclonic epilepsy of infancy, was first described in 1978 and has since become one of the most recognized genetic epilepsy syndromes [42]. The clinical presentation often begins with seizures in the first year of life, frequently in the setting of febrile illness. The initial seizures are generalized or hemiclonic in the majority and are often prolonged evolving to status epilepticus. Unlike typical febrile seizures, one should suspect Dravet syndrome in children that present with repetitive bouts of complex febrile seizures or febrile status epilepticus, especially if the associated seizure semiology is of hemiclonic type. In addition, seizures in the setting of modest hyperthermia (ie, hot baths) should raise suspicion for this condition. Commonly EEG and MRI are normal in the first year of life and psychomotor development remains normal until typically the second year of life [43].
By the second year, other seizure types including myoclonic, atypical absence, clonic, and tonic seizures arise. The EEG frequently begins to show generalized spike wave and polyspike wave discharges. Seizures continue occurring frequently during early childhood, often resulting in status epilepticus. Cognitive development begins to stagnate between the ages of 1 and 4 years with emergence of autistic traits and hyperactivity [44]. Development may stabilize between the ages of 5 and 16 years, but fails to demonstrate much improvement [44]. Higher frequency of seizures may correlate with increase in cognitive impairment and behavior problems, supporting the need for rapid diagnosis and appropriate therapy [44].
Over the years, several cases of atypical or borderline Dravet syndrome have been described, most highlighting the absence of myoclonic seizures [45]. Others may present with primarily clonic or tonic-clonic type seizures only [46]. Despite these differences, all cases share a similar drug resistance and cognitive delay and are categorized as Dravet syndrome.