Hairy Cell Leukemia






Introduction
Hairy cell leukemia (HCL) is a rare chronic lymphoproliferative disorder, with only approximately 2000 new cases diagnosed in the United States each year.1 It is now recognized that there are 2 distinct categories of HCL, classic HCL (cHCL) and variant HCL (vHCL), with vHCL now classified as a separate entity under the World Health Organization Classification of Hematopoietic Tumors.2 For this reason, the 2 diseases will be discussed separately. However, they do bear many clinical and microscopic similarities and because of this were originally indistinguishable using diagnostic techniques previously available. Even in the modern era using immunophenotypic, molecular, and genetic testing, differentiating between the classic and variant disease subtypes is sometimes difficult.
For cHCL the median age of diagnosis is 55 years, with vHCL occurring in patients who are somewhat older; HCL has been described only in the adult population, with 1 exception.3,4 There is a 4:1 male predominance, and Caucasians are more frequently affected than other ethnic groups. While the cause of the disease remains largely unknown, it has been observed to occur more frequently in farmers and in persons exposed to pesticides and/or herbicides, petroleum products, and ionizing radiation.4 The Institute of Medicine recently updated their position regarding veterans and Agent Orange, stating that there is sufficient evidence of an association between herbicides and chronic lymphoid leukemias (including HCL) to consider these diseases linked to exposure.5 Familial forms have also been described that are associated with specific HLA haplotypes, indicating a possible hereditary component.6 Most likely, a combination of environmental and genetic factors ultimately contributes to the development of HCL.
In recent years enormous progress has been made with respect to new insights into the biology of cHCL and vHCL, with significant refinement of diagnostic criteria. In addition, tremendous advances have occurred in both treatment and supportive care regimens, which have resulted in a dramatically increased overall life expectancy as well as decreased disease-related morbidity. This has meant that more patients are affected by HCL over time and are more likely to require care for relapsed HCL or associated comorbidities. Although no curative treatment options exist outside of allogeneic transplantation, therapeutic improvements have resulted in patients with cHCL having a life expectancy similar to that of unaffected patients, increasing the need for vigilance to prevent foreseeable complications.
,Biology and Patheogenisis
The family of HCLs are chronic B-cell malignancies that account for approximately 2% of all diagnosed leukemias.7 The first detailed characterization of HCL as a distinct clinical entity was performed by Dr. Bouroncle and colleagues at the Ohio State University in 1958.8 Originally called leukemic reticuloendotheliosis, it was renamed HCL following more detailed description of the unique morphology of these malignant cells.9 Significant advances have recently been made in identifying distinctive genetic, immunophenotypic, and morphologic features that distinguish HCL from other B-cell malignancies.
HCL B cells tend to accumulate in the bone marrow, splenic red pulp, and (in some cases) peripheral blood. Unlike other lymphoproliferative disorders, HCL only rarely results in lymphadenopathy. HCL derives its name from the distinct appearance of the malignant hairy cells (Figure). Morphologically, HCL cells are mature, small lymphoid B-cells with a round or oval nucleus and abundant pale blue cytoplasm. Irregular projections of cytoplasm and microvilli give the cells a serrated, “hairy” appearance.10 The biological significance of these fine hair-like projections remains unknown and is an area of ongoing investigation. Gene expression profiling has revealed that HCL B cells are most similar to splenic marginal zone B cells and memory B cells.11–13 A recent analysis of common genetic alterations in HCL suggests that the cell of origin is in fact the hematopoietic stem cell.14 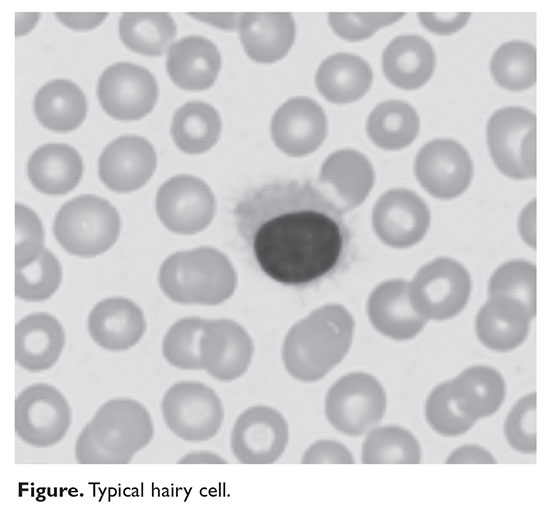
Compared to other hematologic malignancies, the genomic profile of HCL is relatively stable, with few chromosomal defects or translocations observed. A seminal study by Tiacci and colleagues revealed that the BRAF V600E mutation was present in 47 out of 47 cHCL cases examined, results that have since been replicated by other groups, confirming that BRAF V600E is a hallmark mutation in cHCL.15 The BRAF V600E gain-of-function mutation results in constitutive activation of the serine-threonine protein kinase B-Raf, which regulates the mitogen-activated protein kinase (MAPK)/RAF-MEK-ERK pathway. Indeed, cHCL B cells have elevated MAPK signaling, leading to enhancement of growth and survival.16 This specific mutation in the BRAF gene is also seen in a number of solid tumor malignancies including melanoma and thyroid cancer, and represents a therapeutic target using BRAF inhibitors already developed to treat these malignancies.17 Testing for BRAF V600E by polymerase chain reaction or immunohistochemical staining is now routinely performed when HCL is suspected.