Solitary Papule on the Upper Back





THE DIAGNOSIS: Plexiform Palisaded Encapsulated Neuroma
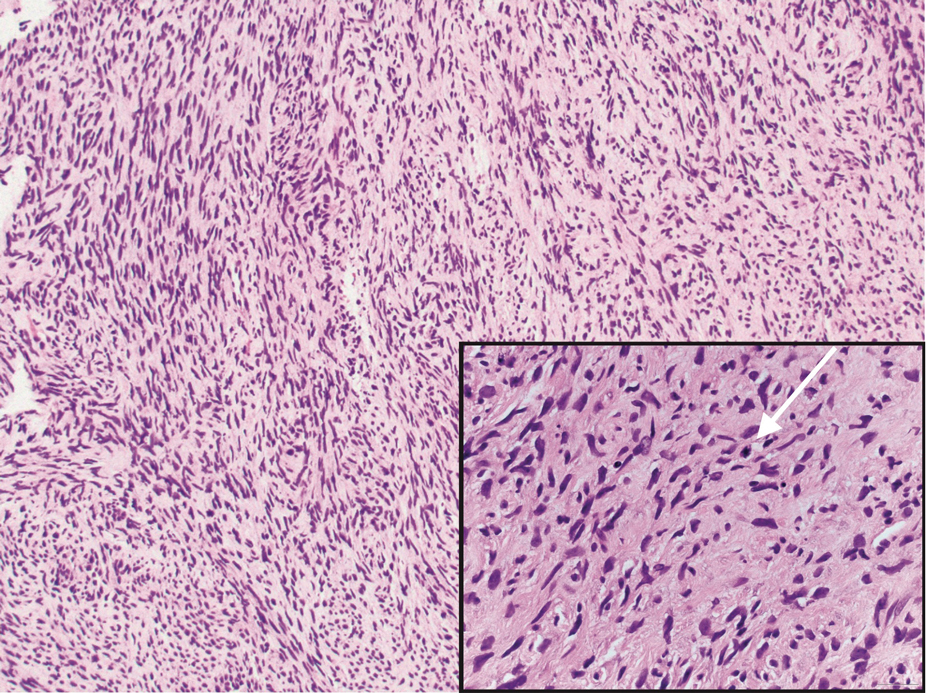
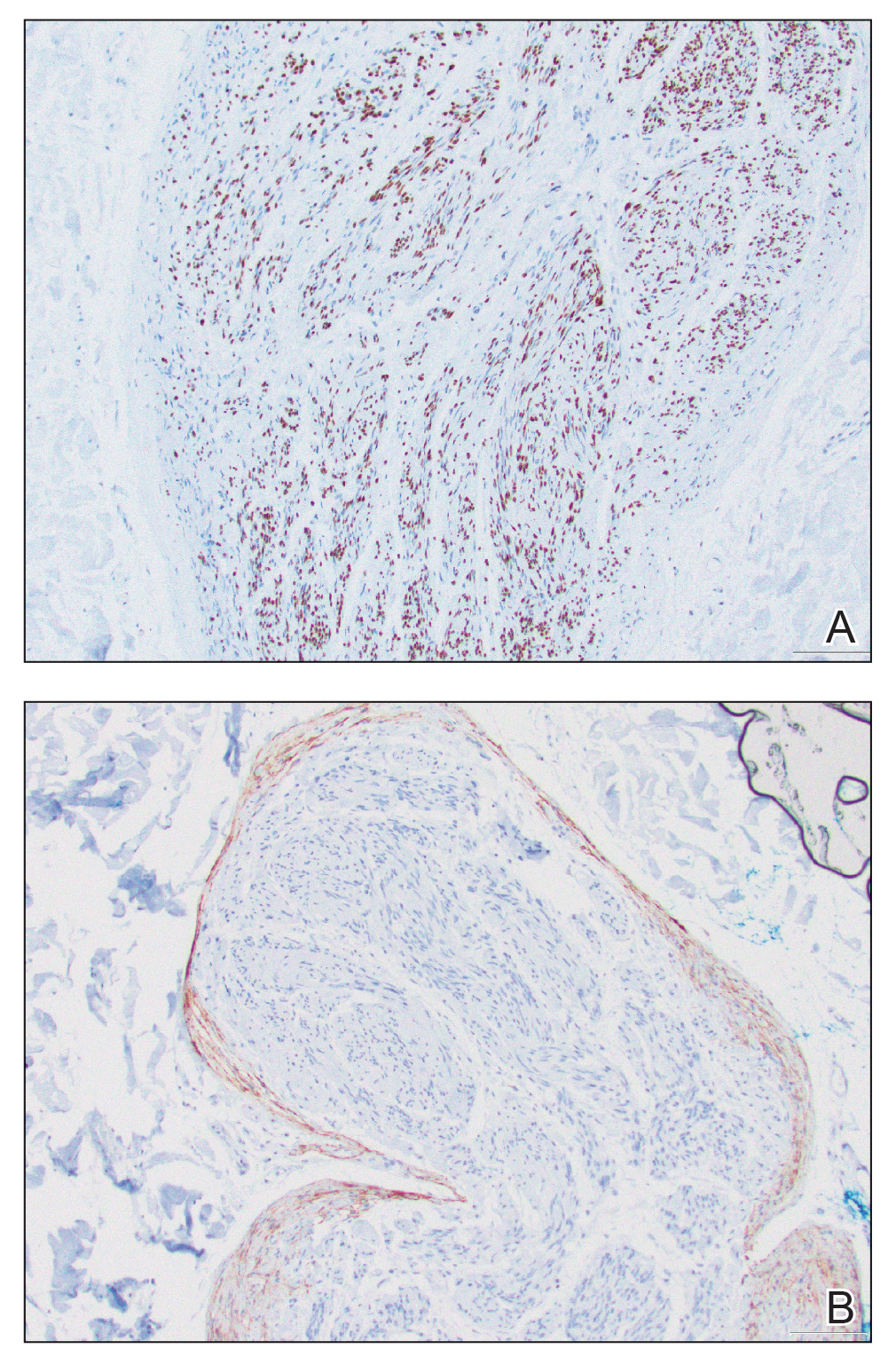
Microscopically, there was a superficial to deep dermal proliferation of tapered spindle cells in fascicles that were well circumscribed in nodules throughout the dermis with pale background stroma, mild mucin, and a thin capsule. The tapered spindle cells stained positive for SOX-10 and negative for Melan-A (Figure 1A). Staining for epithelial membrane antigen highlighted delicate cells around the periphery of the nodules, consistent with perineurium (Figure 1B). A diagnosis of plexiform palisaded encapsulated neuroma was made. No additional treatment was pursued due to the benign nature of the condition.
Palisaded encapsulated neuroma (PEN), also referred to as solitary circumscribed neuroma,1 is a benign, generally solitary neurogenic tumor that manifests predominantly on the skin, particularly in areas of frequent outside trauma such as the face. Lesions also may occur on mucosal and acral sites.2 First described by Reed et al3 in 1972, PEN characteristically manifests as a well-circumscribed, dermal nodule with a distinctive palisading pattern of Schwann cells and axons within a delicate perineurial capsule, the latter of which may be incomplete.3 Palisaded encapsulated neuroma frequently exhibits clefting between the tumor and the surrounding dermis. While PEN generally is sporadic, rare cases have been reported in association with Cowden syndrome and neurofibromatosis type 2.4,5
While the nodular growth pattern is most common, PEN also may present in epithelioid, plexiform, multinodular, or fungating subtypes.6 The plexiform subtype of PEN is rare. It has a complex growth pattern and a tendency to involve multiple adjacent nerve bundles in a plexiform arrangement.6,7 In two independent reviews characterizing the predominant growth patterns of PEN, nonnodular growth patterns were observed in a minority of the 85 cases: 18.8% (16/85) were plexiform, 7.1% (6/85) were multinodular or multilobular, and 7.1% (6/85) were fungating.6,7
The clinical presentation of plexiform PEN often includes a painless, slow-growing mass, and it predominantly occurs in middle-aged adults.2 Immunohistochemical staining reveals diffuse positivity for SOX-10 and S-100, which highlights the neural origin of the tumor.6 This variant, like conventional PEN, lacks notable atypia or mitotic activity.
Palisaded encapsulated neuroma, regardless of subtype, has an excellent prognosis, with no known cases of malignant transformation, and surgical excision with clear margins is curative.8
The differential diagnosis for plexiform PEN includes plexiform variants of neurofibroma and schwannoma, traumatic neuroma, and malignant peripheral nerve sheath tumor.
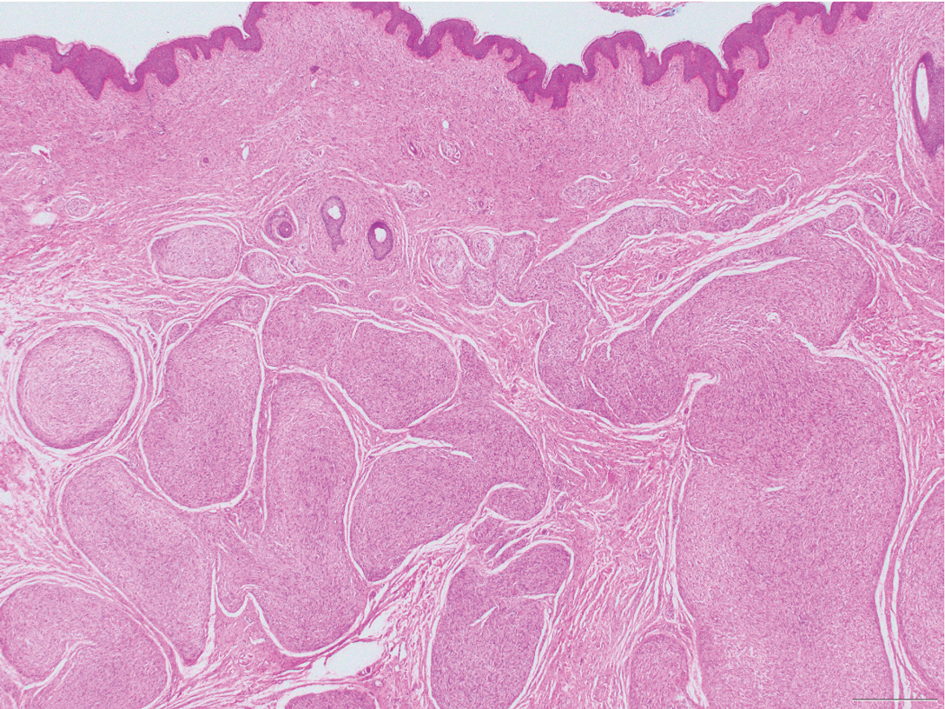
Neurofibromas are nonencapsulated lesions composed of spindle cells with wavy nuclei dispersed in a myxoid background.8 Neurofibromas can manifest in various locations throughout the body, including the skin, subcutaneous tissues, and internal organs. They are slow-growing tumors but may accelerate during periods of hormonal changes, such as pregnancy and puberty, or in cases of malignant transformation.8 Although plexiform neurofibromas are benign, malignant transformation can occur, particularly in patients with neurofibromatosis type 1 (NF1).8,9 Neurofibromas may assume one of 3 growth patterns: localized, diffuse, or plexiform.8 Plexiform neurofibromas exhibit a multinodular, ropelike growth pattern with a mix of Schwann cells and fibroblasts (Figure 2).8,9 These lesions are pathognomonic for NF1 and can infiltrate the surrounding tissue. They may involve large nerve trunks, leading to a more complex growth pattern compared to solitary neurofibromas.8,9 The plexiform variants of both neurofibromas and PEN demonstrate a multinodular growth pattern; however, plexiform neurofibromas are nonencapsulated and show a more diffuse infiltrative nature, whereas plexiform PEN remains well circumscribed. Additionally, plexiform neurofibromas are associated with NF1, while plexiform PEN lacks this genetic association.
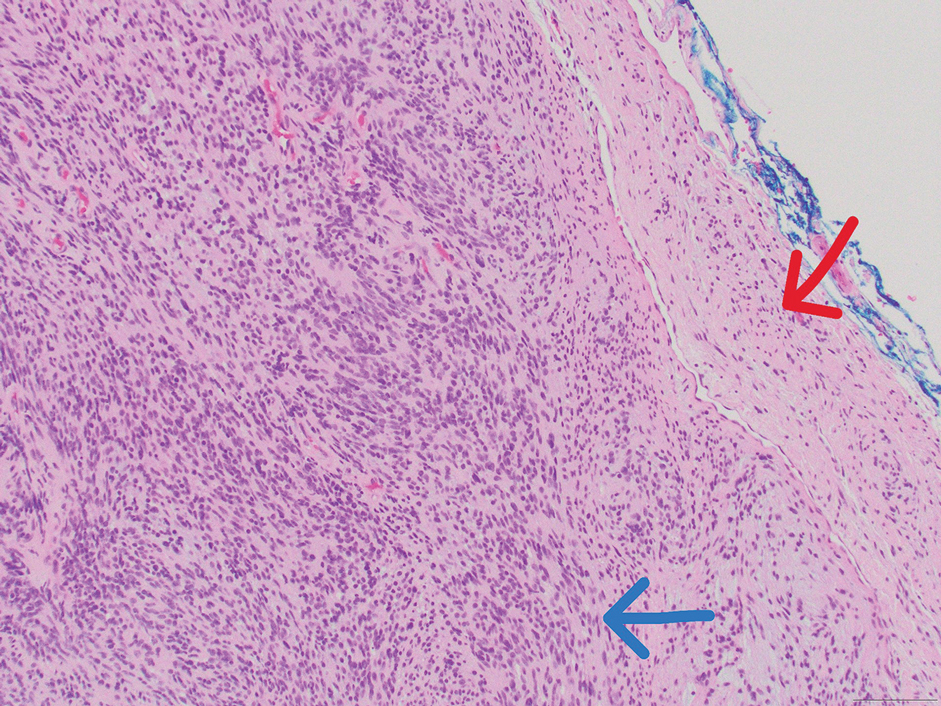
Schwannomas are encapsulated tumors that originate from the outer sheath of peripheral nerves, usually positioned eccentrically to the nerve fibers. Schwannomas are characterized by Antoni A and Antoni B areas, which usually are absent in PEN. Antoni A areas are composed of compact spindle cells arranged in palisades with Verocay bodies, while Antoni B areas are more loosely arranged and have a myxoid background (Figure 3).8,9 Schwannomas stain positive for S-100 and often show degenerative changes such as cystic degeneration or calcification, particularly in larger lesions.8,9 Plexiform schwannoma is a rare variant of schwannoma, and while it carries a substantial risk for local recurrence with rates as high as 50%, it has not been shown to possess malignant or metastatic potential.10 Unlike PEN, schwannomas have a consistent capsule but share S-100 positivity with PEN. Verocay bodies occasionally can be observed in PENs, with studies reporting their presence in 20% to 36% of cases.7,11,12 Additionally, some schwannomas may exhibit few Verocay bodies or poorly developed forms, which can make histopathologic distinction more challenging.7,11,12
Traumatic neuromas result from nerve regeneration following any type of outside trauma that is deep enough to cause nerve injury. The lesion often is painful and associated with prior trauma or surgery. Under optimal conditions, the severed ends of a nerve reconnect through the orderly growth of axons from the proximal stump to the distal stump, guided by tubes formed by proliferating Schwann cells. If the nerve ends are not properly aligned or if the distal stump is absent, the axons may proliferate in a disorganized manner at the proximal stump, resulting in the formation of a traumatic neuroma.8 Histologically, these lesions exhibit disorganized, proliferating nerve fibers intermixed with fibrous stroma.8,13 The nerve fibers are not encapsulated, and there is an irregular arrangement of axons and Schwann cells (Figure 4).8,13 Unlike PEN, which usually is encapsulated and well organized with fascicular architecture, traumatic neuromas exhibit a disorganized, haphazard arrangement of neural elements and lack a capsule.8 Clinically, traumatic neuromas also are more likely to be painful.
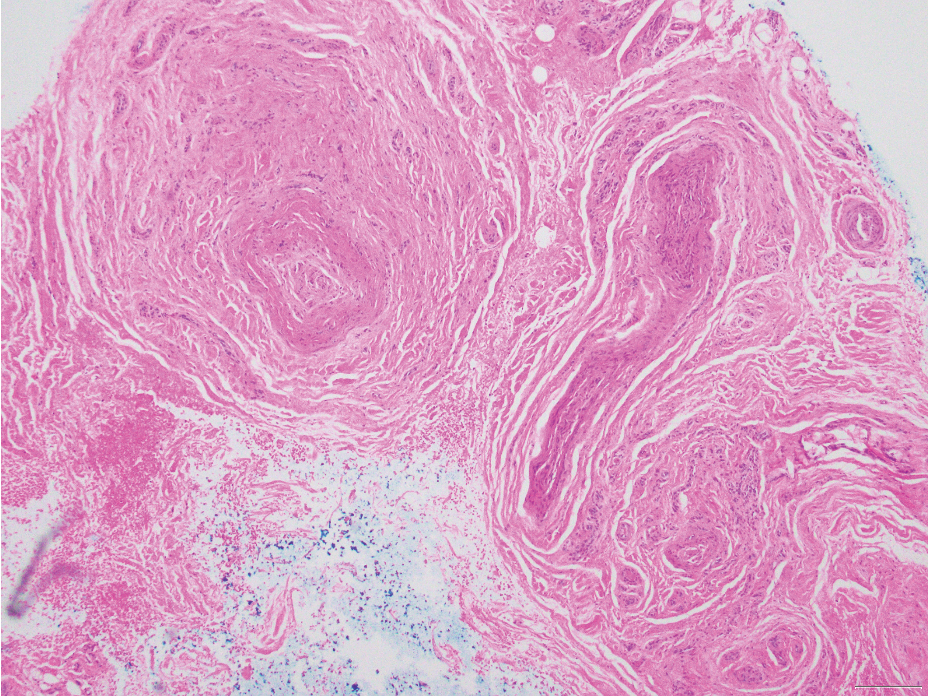
Malignant peripheral nerve sheath tumors are aggressive malignant spindle-cell tumors that may be associated with NF1 or occur sporadically.9,14 The spindle cells are arranged in fascicles, and these tumors can have areas of necrosis, hemorrhage, and high mitotic activity.9,15 The spindle cells may be arranged in a herringbone pattern, and alternating areas of hypocellularity and hypercellularity impart a marbled appearance (Figure 5).16 Malignant peripheral nerve sheath tumors frequently exhibit inactivation of the SWI/SNF-related, matrix- associated, actin-dependent regulator of chromatin subfamily B member 1 gene and loss of integrase interactor 1 protein. Transformation from plexiform neurofibroma to malignant peripheral nerve sheath tumor frequently is accompanied by progressive genomic changes.17 Malignant peripheral nerve sheath tumors differ substantially from PEN in their aggressive histologic features, including nuclear atypia and mitotic figures, which are absent in PEN.