Purpuric Macule of the Right Axilla





The Diagnosis: Atypical Vascular Lesion
Atypical vascular lesion (AVL)(quiz image), named by Fineberg and Rosen,1 is a vascular lesion that arises on mammary skin with a history of radiation exposure. Clinically, AVL can present as a papule or erythematous patch that manifests 3 to 7 years after radiation therapy.2,3 There are 2 histologic subtypes of AVL: lymphatic and vascular.2,4 Lymphatic-type AVL is comprised of a symmetric distribution of thin, dilated, and anastomosing vessels usually found in the superficial and mid dermis. The vessels are lined by flat or hobnail protuberant endothelial cells that lack nuclear irregularity or pleomorphism; however, hyperchromatism of endothelial cell nuclei is a common finding. Vascular-type AVL is morphologically similar to a capillary hemangioma, and histologic features include irregular growth of capillary-sized vessels that extend to the dermis and subcutis.2,4 Atypical vascular lesions are benign lesions but may be a precursor to angiosarcoma. Along with vascular markers, D2-40 typically is positive. Surgical excision with clear margins is recommended when the lesion is small.4,5 Observation is more appropriate for extensive lesions.
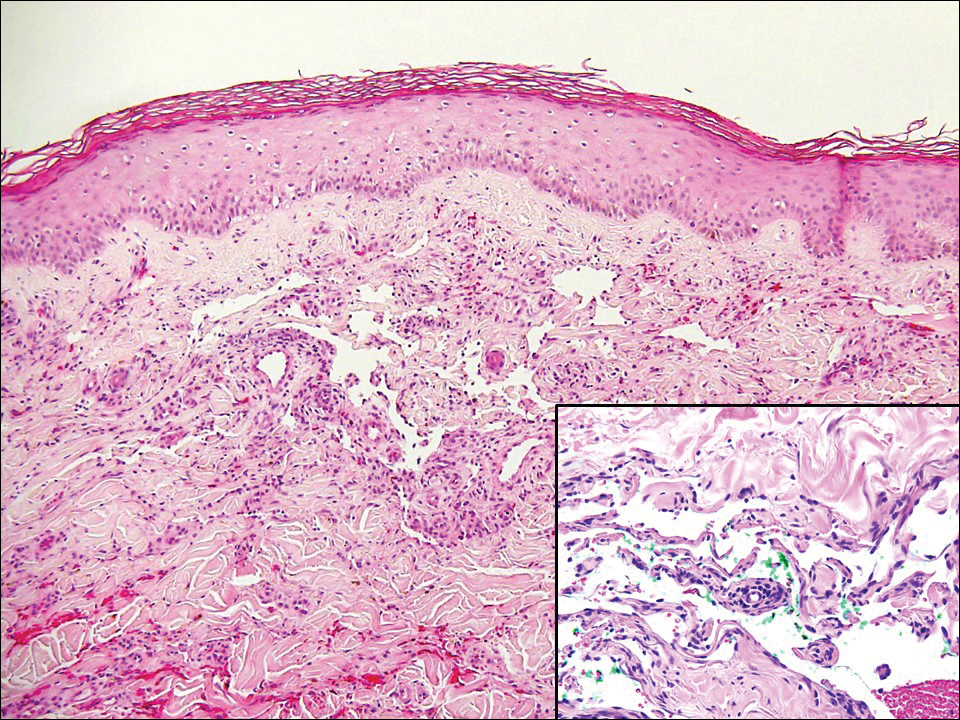
Angiosarcoma can arise spontaneously or in association with radiation or chronic lymphedema. Given the shared risk factors and presentation with AVL, it is essential to differentiate angiosarcoma from AVL. Primary cutaneous angiosarcoma usually presents on the head of elderly patients as an ecchymotic patch or plaque with ulceration.4 Secondary angiosarcoma may arise following radiation or chronic lymphedema (Stewart-Treves syndrome); however, some authors now prefer to consider lymphangiosarcoma arising in chronic lymphedematous limbs a distinct entity.6 Surgical excision with wide margins is the mainstay of therapy, but angiosarcoma has high recurrence rates, and the 5-year survival rate has been reported to be as low as 35%.7 Histologic overlap with AVL includes dissecting anastomosing vessels lined by hyperchromatic nuclei; however, angiosarcoma is distinguished by endothelial cell layering, nuclear pleomorphism, and prominent nucleoli (Figure 1).4,8 Increased positivity for Ki-67 immunostain, which indicates cell proliferation, may be used to distinguish angiosarcoma from an AVL (Figure 1 [inset]).9 Further, in contrast to AVL, radiation-induced angiosarcoma is characterized by amplification of C-MYC, a regulator gene, and FLT4 (FMS-related tyrosine kinase 4), a gene encoding vascular endothelial growth factor receptor 3. Gene amplification may be detected through immunohistochemistry or fluorescence in situ hybridization.10 Ki-67 labeling showed less than 10% staining in endothelial cells in our case (quiz image [inset]), and fluorescence in situ hybridization was negative for C-MYC amplification, supporting the diagnosis of AVL.
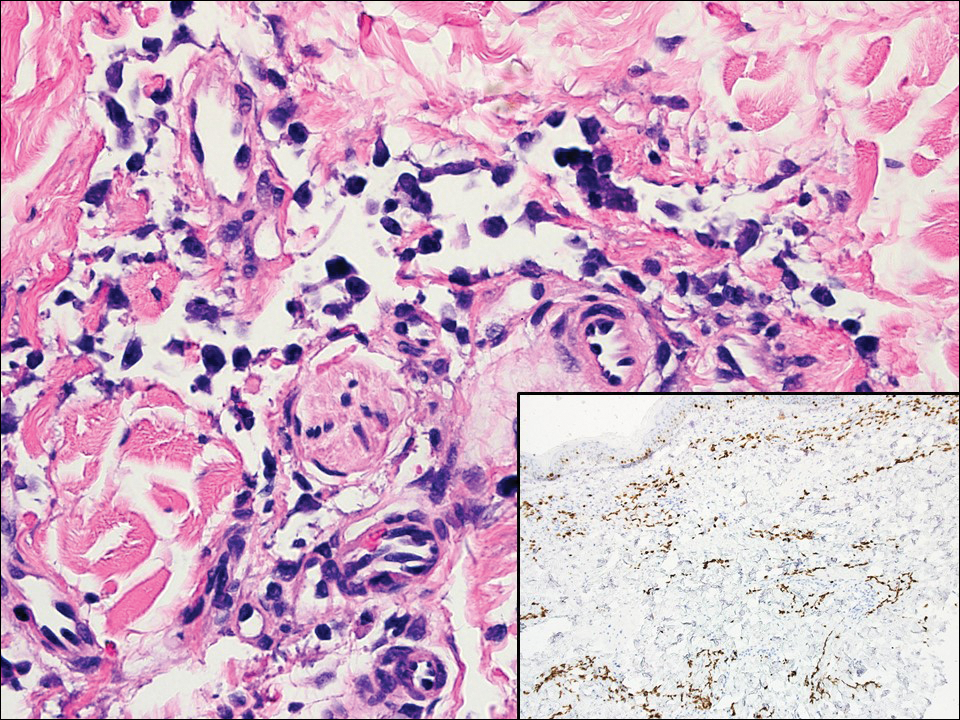
Lymphangioma circumscriptum, the most common superficial lymphangioma, is a hamartomatous malformation that usually occurs at the axillary folds, neck, and trunk. It clinically presents as small agminated vesicles with a characteristic frog spawn appearance.11 Dermoscopic features include yellow lacunae that may alternate with a dark red color secondary to extravasation of erythrocytes.12 These clinical features often lead to a differential diagnosis of verrucae, angiokeratoma, and angiosarcoma. Lymphangioma circumscriptum histologically is characterized by an overgrowth of dilated lymphatic vessels that fill the papillary dermis. The vessels are composed of flat endothelial cells typically filled with acellular proteinaceous debris and occasional erythrocytes (Figure 2). As the lesion traverses deeper into the dermis, the caliber of the lymphatic channel becomes narrower. The presence of deep lymphatic cisterns with surrounding smooth muscle is helpful to differentiate lymphangioma circumscriptum from other lymphatic malformations such as acquired lymphangiectasia. Treatment options include surgical excision, sclerosing agents, and destructive modalities such as cryotherapy.
,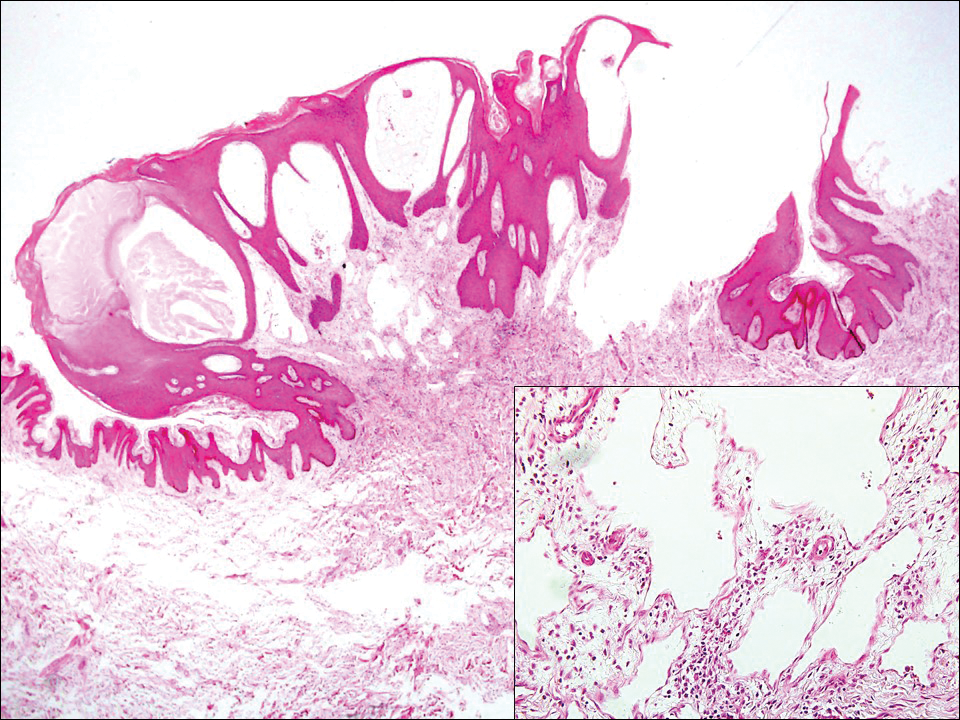
Hobnail hemangioma, originally termed targetoid hemosiderotic hemangioma by Santa Cruz and Aronberg,13 presents as a violaceous papule or nodule surrounded by a characteristic brown halo on the leg. Trauma has been proposed as the inciting factor for the clinical appearance of hobnail hemangioma.14 Microscopically, the lesion shows vessels in a wedge shape. The superficial component has telangiectatic vessels with focal areas of papillary projections lined by endothelial cells. Although the endothelial nuclei typically project into the lumen, the nuclei are small, bland, and without mitotic activity.15 Deeper components show slit-shaped vasculature with dermal collagen dissection. Hemosiderin, extravasated red blood cells, and inflammation are found adjacent to the vessels (Figure 3). Given the benign nature, hobnail hemangiomas may be monitored.
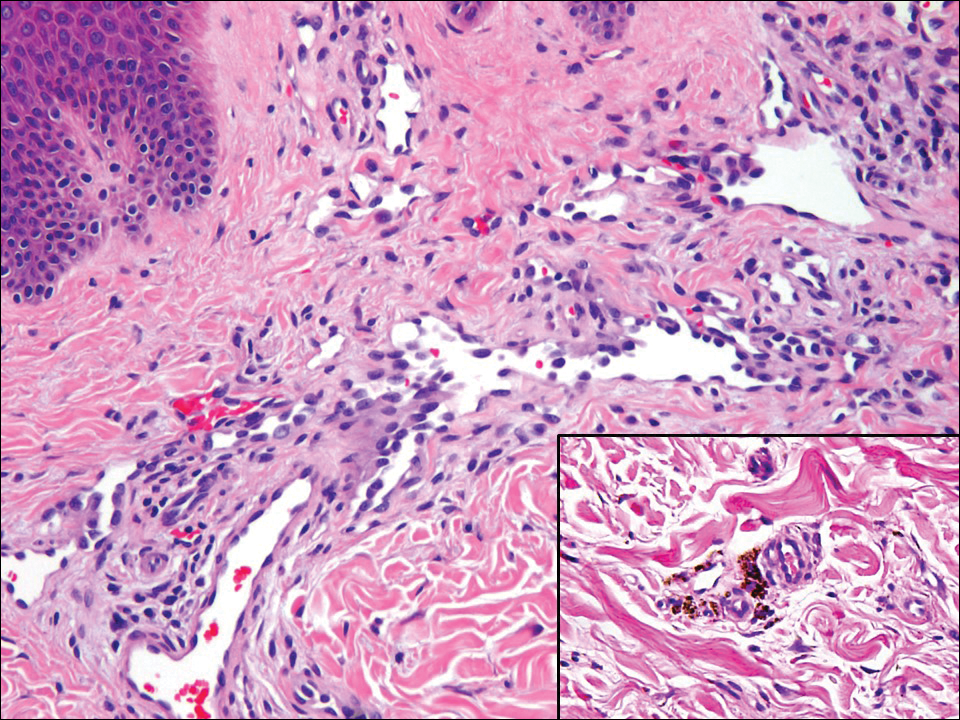
Kaposi sarcoma (KS) is a low-grade vascular neoplasm associated with human herpesvirus 8 that arises in multiple clinical settings, especially in immunosuppression secondary to human immunodeficiency virus. There are 3 distinct clinical stages: patch, plaque, and tumor. The patch stage appears as red macules that blend into larger plaques; the tumor stage is defined as larger nodules developing from plaques. Histologic features differ by stage. Similar to angiosarcoma, KS is comprised of anastomosing vessels that dissect collagen bundles; endothelial cell atypia is minimal. A useful feature of KS is its propensity to involve adnexa and display the promontory sign, which involves the tumor growing into normal vasculature (Figure 4).16 Positive immunohistochemistry for human herpesvirus 8 aids in confirmation of the diagnosis. Treatment options for KS are numerous but include destructive modalities, chemotherapeutic agents such as doxorubicin, or highly active antiretroviral therapy for AIDS-related KS.17