Richner-Hanhart Syndrome (Tyrosinemia Type II)





Practice Points
- Richner-Hanhart syndrome (tyrosinemia type II) should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.
- Early diagnosis and initiation of a tyrosinephenylalanine–restricted diet in infancy is the most effective therapy to prevent mental retardation.
To the Editor:
Richner-Hanhart syndrome, also known as tyrosinemia type II or oculocutaneous tyrosinemia, is a rare autosomal-recessive, childhood-onset, metabolic hereditary disease.1 A deficiency of tyrosine aminotransferase leads to an accumulation of tyrosine amino acid. It is characterized by the association of palmoplantar hyperkeratosis, bilateral keratitis, and neurological disorders.
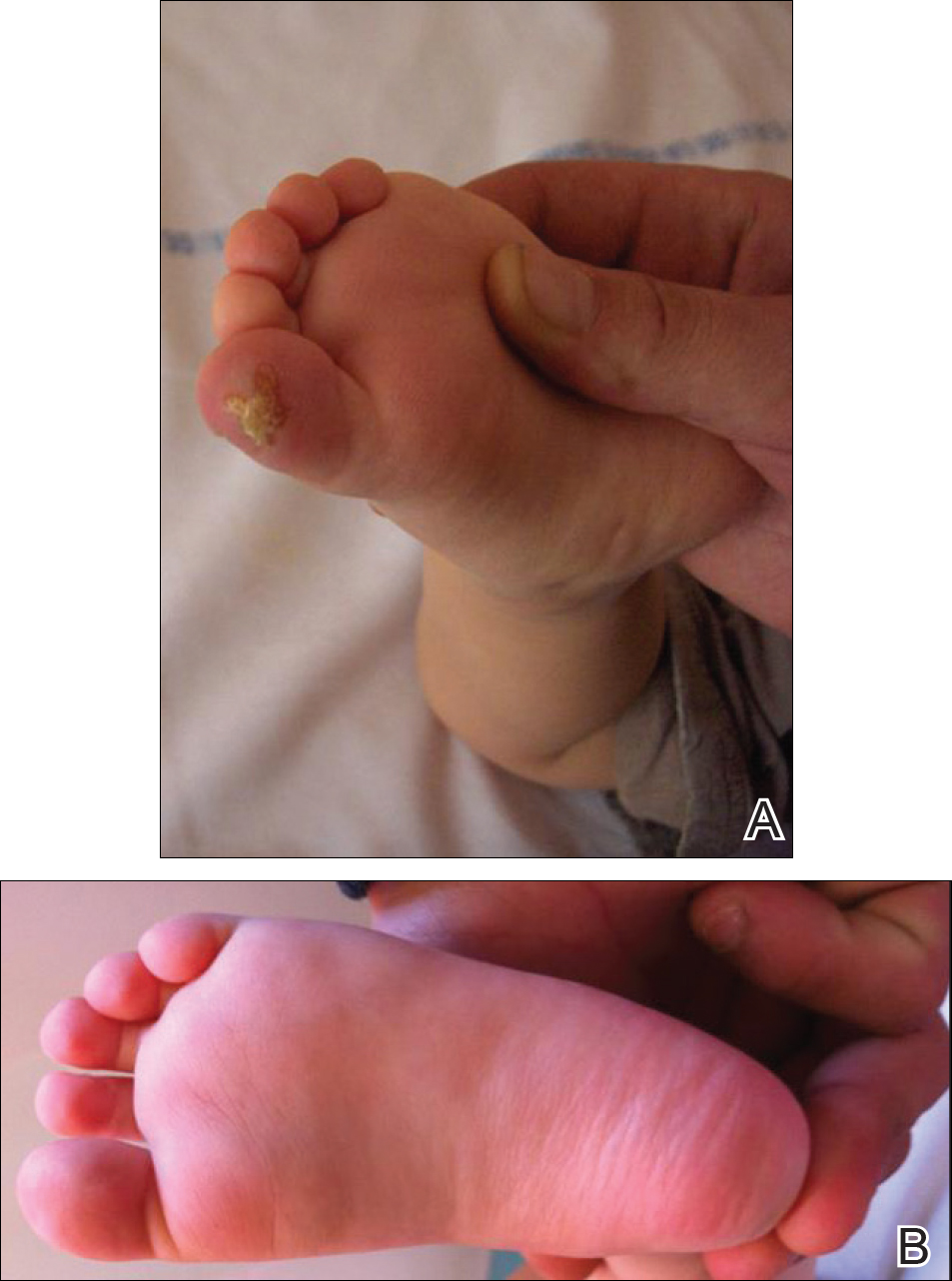
An 18-month-old girl with recurrent warts of 6 months' duration was admitted to the dermatology department. She had been treated repeatedly with acyclovir for recurrent bilateral herpetic keratitis with major photophobia since 9 months of age with no response. Clinical presentation included punctate hyperkeratosis of the fingers and toes (Figure, A), severe photophobia with decreased visual acuity, and speech delay.
,
Her medical record showed a break of the growth curve with a weight of 9.25 kg (3rd percentile), a height of 80 cm (50th percentile), and a head circumference of 45 cm (50th percentile). Her parents were nonconsanguineous. The association of bilateral dendritic keratitis with punctate palmoplantar keratosis suggested a diagnosis of Richner-Hanhart syndrome. Diagnosis was confirmed by an elevated plasma level of tyrosine (1580 µmol/L; reference range, 40-80 µmol/L).
A low tyrosine and low phenylalanine diet (no animal proteins) was immediately introduced, with supplementation of amino acids, vitamins, and trace elements. After 8 days, the plasma level of tyrosinemia decreased by a factor of 4 (392 µmol/L). After 1 month, the cutaneous and ocular lesions completely resolved (Figure, B). Discrete psychomotor slowing still persisted for 1 year and then reached complete normalization. Genetic analysis showed a composite heterozygous mutation of the tyrosine aminotransferase gene, TAT, on chromosome 16. The mutation detected in the patient's mother was an A to V substitution at codon 147 (A147V). The second mutation was detected in the father; it was an 8 nucleotides duplication and then a substitution leading to a premature stop codon at codon 37 (R37X).
Richner-Hanhart syndrome is a rare autosomal-recessive disorder that is more common in Italy and in areas where inbreeding is prevalent1,2; however, no data are available on disease prevalence. It is caused by a homozygous mutation in the TAT gene located on chromosome 16q22.3 Tyrosine aminotransferase is an important enzyme involved in the tyrosine and phenylalanine metabolic degradation pathway located in the hepatic cytosol. Symptoms are due to the accumulation of tyrosine and its metabolite. Diagnosis is confirmed by an elevated plasma level of tyrosine (>500 µmol/L). This oculocutaneous syndrome is characterized by bilateral pseudodendritic keratitis, palmoplantar hyperkeratosis, and a variable degree of mental retardation.1 In contrast to tyrosinemia type II, types I and III do not affect the skin.
Intrafamilial and interfamilial phenotypic variability is reported. A large spectrum of mutations within the TAT gene have been reported.4-7 These mutations lead to a reduction or an absence in the activity of hepatic tyrosine aminotransferase. The degradation pathway of tyrosine involving TAT occurs mainly in the liver. This process also is present in the mitochondria where the enzyme is called aspartate aminotransferase.1,2 The mechanism by which Richner-Hanhart syndrome causes painful palmoplantar keratosis and keratitis remains unknown. It has been suggested that intracellular L-tyrosine crystals initiate an inflammation process resulting in the typical skin lesions and keratitis.8 There is some evidence that patients with higher values of tyrosine in early life are more likely to develop neurological problems.1 In addition, phenotype variability has been observed, even among individuals sharing the same pathogenic mutation.4
Tyrosinemia type II typically demonstrates ocular symptoms (75% of cases) that usually occur in the first year of life.8 They are characterized by photophobia, redness, and increase of lacrimation. Examination reveals a superficial and bilateral punctate keratosis with corneal dystrophy, often misdiagnosed as herpetic keratosis, as in our case, which may delay the diagnosis.9,10 Bilateral ocular lesions are suggestive, even if they are asymmetric.8,11 Furthermore, negative fluorescein staining, negative culture, and resistance to antiviral treatment exclude the diagnosis of herpetic keratosis.9,10
Skin lesions (85% of cases) typically appear in the first year of life. They are characterized by painful, irregular, limited, punctate hyperkeratosis on the palms and soles.1 They are more frequent in weight-bearing areas and tend to improve during summer, possibly due to a seasonal change in dietary behavior.4,12 Hyperkeratotic papules in a linear pattern also have been described on the flexor aspects of the fingers or toes.13 In our case, the lesions were misdiagnosed as warts for 6 months.
Retarded development affects 60% of patients with tyrosinemia type II. Expression of neurological symptoms is variable and could include mental retardation, nystagmus, tremors, ataxia, and convulsion.4 Lifetime follow-up of these patients is recommended.
Early initiation of a tyrosine-phenylalanine-restricted diet in infancy is the most effective therapy for Richner-Hanhart syndrome.13 The enzyme phenylalanine hydroxylase normally converts the amino acid phenylalanine into amino acid tyrosine. Thus, dietary treatment of Richner-Hanhart syndrome requires restricting or eliminating foods high in phenylalanine and tyrosine with protein "medical food" substitute. The dietary treatment allows resolution of both eye and skin symptoms after a few days or weeks and also may prevent mental retardation. It is effective in lowering the plasma level to less than 400 µmol/L. The diet must be introduced as soon as Richner-Hanhart syndrome is suspected. Supplementation with essential amino acids, vitamins, and trace elements is needed. Early screening of siblings in families with Richner-Hanhart syndrome history is recommended, even in the absence of clinical findings. Careful dietary control of maternal plasma tyrosine level must be considered during future pregnancy for women.4,14,15
Richner-Hanhart syndrome should be suspected in patients demonstrating cutaneous lesions, especially palmoplantar keratosis associated with bilateral pseudodendritic corneal lesions unresponsive to antiviral therapy.