Cystic lung disease: Systematic, stepwise diagnosis





ABSTRACTOnce cystic lung disease is confirmed on computed tomography, one can arrive at the likely diagnosis in most cases by taking a systematic, stepwise approach based on the clinical and radiographic features. Here, we describe the features of cystic lung disease that point to lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary angerhans cell histiocytosis, interstitial pneumonia, congenital cystic lung disease, pulmonary infection, and systemic disease.
KEY POINTS
- Pulmonary cysts should be differentiated from cyst-mimics.
- Adults with cystic lung disease can be grouped by the clinical presentation: ie, insidious dyspnea or spontaneous pneumothorax; incidentally found cysts or recurrent pneumonia; signs and symptoms of primary pulmonary infection; or signs and symptoms that are primarily nonpulmonary.
- Characterization of pulmonary cysts and their distribution plays a key role in diagnosis. Radiographically, cystic lung disease can be subclassified into two major categories according to the distribution of cysts: discrete (focal or multifocal) and diffuse (unilobular or panlobular).
Primarily nonpulmonary signs and symptoms
If the patient has primarily nonpulmonary signs and symptoms, think about pulmonary amyloidosis, light chain deposition disease, and neurofibromatosis type 1.
Pulmonary amyloidosis has a variety of manifestations, including tracheobronchial disease, nodular parenchymal disease, diffuse or alveolar septal pattern, pleural disease, lymphadenopathy, and pulmonary cysts.4
Light chain deposition disease shares some clinical features with amyloidosis. However, the light chain fragments in this disease do not form amyloid fibrils and therefore do not stain positively with Congo red. The kidney is the most commonly involved organ.4
Neurofibromatosis type 1 is characterized by collections of neurofibromas, café-au-lait spots, and pigmented hamartomas in the iris (Lisch nodules).35
In summary, patients in this group typically present with complications related to systemic involvement. Those with neurofibromatosis type 1 present with ophthalmologic, dermatologic, and neurologic manifestations. Amyloidosis and light chain deposition disease most commonly involve the renal system; their distinction will likely require tissue biopsy and Congo-red staining.
STEP 3: CHARACTERIZE THE RADIOGRAPHIC FEATURES
Characterization of pulmonary cysts and their distribution plays a key role in the diagnosis. Radiographically, cystic lung diseases can be subclassified into two major categories according to their cystic distribution:
- Discrete (focal or multifocal)
- Diffuse (unilobular or panlobular).2,3
Discrete cystic lung diseases include congenital abnormalities, infectious diseases, and interstitial pneumonias.2,3
Diffuse, panlobular cystic lung diseases include lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, Birt-Hogg-Dubé syndrome, amyloidosis, light chain deposition disease, and neurofibromatosis type 1.7,13,36–39
In addition, other associated radiographic findings play a major role in diagnosis.
Cysts in patients presenting with insidious dyspnea or spontaneous pneumothorax
Lymphangioleiomyomatosis. Cysts are seen in nearly all cases of advanced lymphangioleiomyomatosis, typically in a diffuse pattern, varying from 2 mm to 40 mm in diameter, and uniform in shape (Figure 2A).7,8,40–42
Other radiographic features include vessels located at the periphery of the cysts (in contrast to the centrilobular pattern seen with emphysema), and chylous pleural effusions (in about 22% of patients).40 Nodules are typically not seen with lymphangioleiomyomatosis, and if found represent type 2 pneumocyte hyperplasia.
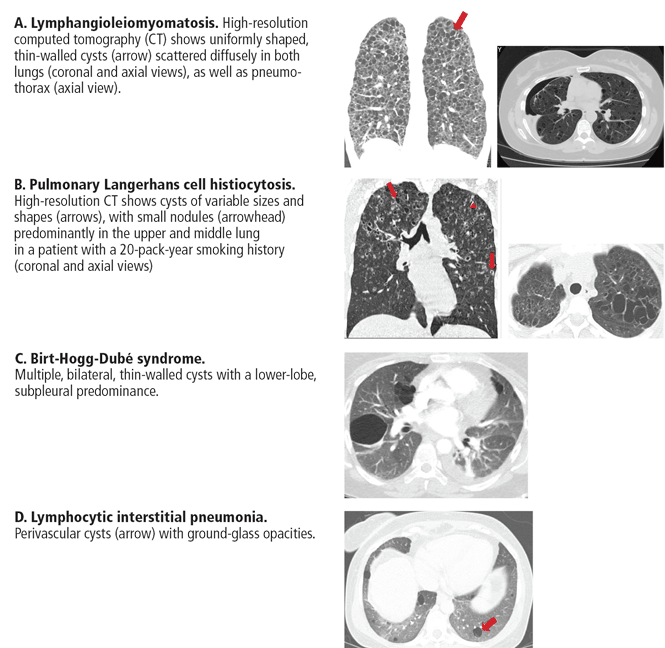
Pulmonary Langerhans cell histiocytosis. Nodules measuring 1 to 10 mm in diameter and favoring a centrilobular location are often seen on computed tomography. Pulmonary cysts occur in about 61% of patients.13,43 Cysts are variable in size and shape (Figure 2B), in contrast to their uniform appearance in lymphangioleiomyomatosis. Most cysts are less than 10 mm in diameter; however, they can be up to 80 mm.13,43 Early in its course, nodules may predominate in the upper and middle lobes. Over time, diffuse cysts become more common and can be difficult to differentiate from advanced smoking-induced emphysema.44
Birt-Hogg-Dubé syndrome. Approximately 70% to 100% of patients with Birt-Hogg-Dubé syndrome will have multiple pulmonary cysts detected on computed tomography. These cysts are characteristically basal and subpleural in location, with varying sizes and irregular shapes in otherwise normal lung parenchyma (Figure 2C).36,45,46
Desquamative interstitial pneumonia. Pulmonary cysts are present on computed tomography in about 32% of patients.47 They are usually round and less than 20 mm in diameter.48 Ground-glass opacity is present in almost all cases of desquamative interstitial pneumonia, with a diffuse pattern in 25% to 44% of patients.16,17,47
Pulmonary cysts occur in up to two-thirds of those with lymphocytic interstitial pneumonia. Cysts are usually multifocal and perivascular in distribution and have varying sizes and shapes (Figure 2D).22 Ground-glass opacity and poorly defined centrilobular nodules are also frequently seen. Other computed tomographic findings include thickening of the bronchovascular bundles, focal consolidation, interseptal lobular thickening, pleural thickening, and lymph node enlargement.22
In summary, in this group of patients, diffuse panlobular cysts are due to lymphangioleiomyomatosis, pulmonary Langerhans cell histiocytosis, or Birt-Hogg-Dubé syndrome. Cysts due to lymphangioleiomyomatosis have a diffuse distribution, while those due to pulmonary Langerhans cell histiocytosis tend to be upper-lobe-predominant and in the early stages are associated with stellate centrilobular nodules. Cysts in Birt-Hogg-Dubé syndrome tend to be subpleural and those due to lymphocytic interstitial pneumonia are perivascular in distribution.
Cysts that are incidentally found or occur in patients with recurrent pneumonia
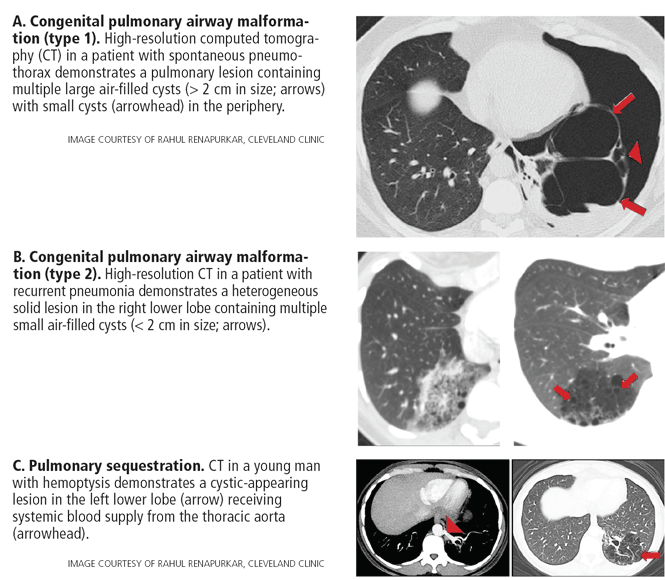
Congenital pulmonary airway malformation types 1, 2, and 4 (Figure 3A, 3B). Cysts are typically discrete and focal or multifocal in distribution, but cases of multilobar and bilateral distribution have also been reported.27,49 The lower lobes are more often involved.49 Cysts vary in size and shape and can contain air, fluid, or both.27,49 Up to 50% of cases can occur in conjunction with pulmonary sequestration.50
Pulmonary sequestration displays an anomalous arterial supply on computed tomography (Figure 3C). Other imaging findings include mass lesions (49%), cystic lesions (29%), cavitary lesions (12%), and bronchiectasis.30 Air trapping can be seen in the adjacent lung. Lower lobe involvement accounts for more than 95% of total cases of sequestration.30 The cysts are usually discrete or focal in distribution. Misdiagnosis of pulmonary sequestration is common, and can include pulmonary abscess, pneumonia, bronchiectasis, and lung cancer.30
Bronchogenic cyst. Cyst contents generally demonstrate water attenuation, or higher attenuation if filled with proteinaceous/mucoid material or calcium deposits; air-fluid levels are seen in infected cysts.32 Intrapulmonary cysts have a predilection for the lower lobes and are usually discrete or focal in distribution.31,32 Mediastinal cysts are usually homogeneous, solitary, and located in the middle mediastinum.32 Cysts vary in size from 20 to 90 mm, with a mean diameter of 40 mm.31
In summary, in this group of cystic lung diseases, characteristic computed tomographic findings will suggest the diagnosis—air-filled cysts of varying sizes for congenital pulmonary airway malformation and anomalous vascular supply for pulmonary sequestration. Bronchogenic cysts will tend to have water or higher-than-water attenuation due to proteinaceous-mucoid material or calcium deposits.
Cysts in patients with signs and symptoms of primary pulmonary infections
P jirovecii pneumonia. Between 10% and 15% of patients have cysts, and about 18% present with spontaneous pneumothorax.51 Cysts in P jirovecii pneumonia vary in size from 15 to 85 mm in diameter and tend to occur in the upper lobes (Figure 4A).51,52
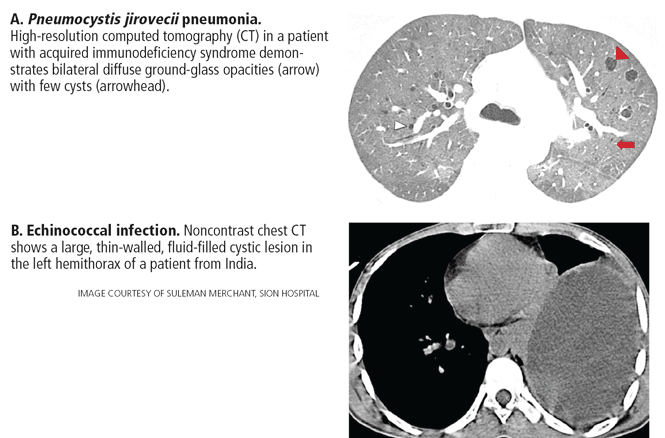
Echinococcal infection. Echinococcal pulmonary cysts typically are single and located more often in the lower lobes (Figure 4B).53,54 Cysts can be complicated by air-fluid levels, hydropneumothorax, or pneumothorax, or they can turn into cavitary lesions.
The diagnoses of these pulmonary infections are usually made by clinical and computed tomographic findings and depend less on detecting and characterizing lung cysts. Patients with P jirovecii pneumonia tend to have bilateral perihilar ground-glass opacities, while air-fluid levels suggest echinococcal infections. Cysts in this group of patients tend to be discrete or focal or multifocal in distribution, and vary in size.
Cysts in patients with primarily nonpulmonary signs and symptoms
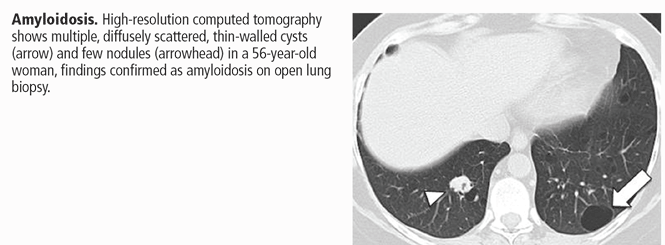
Amyloidosis. Cyst formation is rare in amyloidosis.4 When present, cysts can be diffuse and scattered in distribution, in varying sizes (usually < 30 mm in diameter) and irregular shapes (Figure 5).55,56
Pulmonary light chain deposition disease usually presents as linear opacities and small nodules on chest computed tomography. Numerous cysts that are diffuse in distribution and have no topographic predominance can also be present. They can progress in number and size and coalesce to form irregular shapes.57
Neurofibromatosis type 1. In neurofibromatosis type 1, the most common radiographic presentations are bibasilar reticular opacities (50%), bullae (50%), and ground glass opacities (37%).58 Well-formed cysts occur in up to 25% of patients and tend to be diffuse and smaller (2 to 18 mm in diameter), with upper lobe predominance.58,59
In summary, in this group of patients, bibasilar reticular and ground-glass opacities suggest neurofibromatosis type 1, while nodules and linear opacities suggest amyloidosis or light chain deposition disease. Cysts tend to be diffuse with varying sizes.