Cystic lung disease: Systematic, stepwise diagnosis





ABSTRACTOnce cystic lung disease is confirmed on computed tomography, one can arrive at the likely diagnosis in most cases by taking a systematic, stepwise approach based on the clinical and radiographic features. Here, we describe the features of cystic lung disease that point to lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary angerhans cell histiocytosis, interstitial pneumonia, congenital cystic lung disease, pulmonary infection, and systemic disease.
KEY POINTS
- Pulmonary cysts should be differentiated from cyst-mimics.
- Adults with cystic lung disease can be grouped by the clinical presentation: ie, insidious dyspnea or spontaneous pneumothorax; incidentally found cysts or recurrent pneumonia; signs and symptoms of primary pulmonary infection; or signs and symptoms that are primarily nonpulmonary.
- Characterization of pulmonary cysts and their distribution plays a key role in diagnosis. Radiographically, cystic lung disease can be subclassified into two major categories according to the distribution of cysts: discrete (focal or multifocal) and diffuse (unilobular or panlobular).
STEP 2: CHARACTERIZE THE CLINICAL PRESENTATION
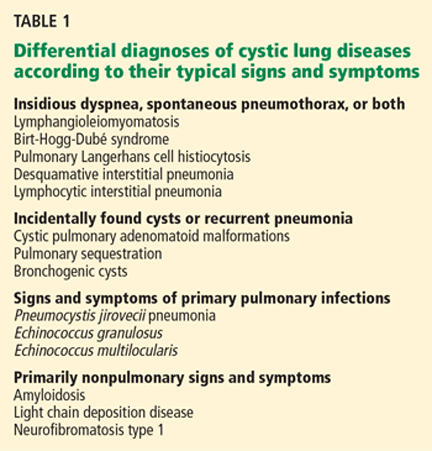
Clinical signs and symptoms of cystic lung disease play a key role in diagnosis (Table 1). For instance, spontaneous pneumothorax is commonly associated with diffuse cystic lung disease (lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome), while insidious dyspnea, with or without associated pneumothorax, is usually associated with the interstitial pneumonias (lymphocytic interstitial pneumonia and desquamative interstitial pneumonia).
In addition, congenital abnormalities of the lung can lead to cyst formation. These abnormalities, especially when associated with other congenital abnormalities, are often diagnosed in the prenatal and perinatal periods. However, some remain undetected until incidentally found later in adulthood or if superimposing infection develops.
Primary pulmonary infections can also cause parenchymal necrosis, which in turn cavitates or forms cysts.4
Lastly, cystic lung diseases can occur as part of a multiorgan or systemic illness in which the lung is one of the organs involved. Although usually diagnosed before the discovery of cysts or manifestations of pulmonary symptoms, they can present as a diagnostic challenge, especially when lung cysts are the initial presentation.bsence of amyloid fibrils.
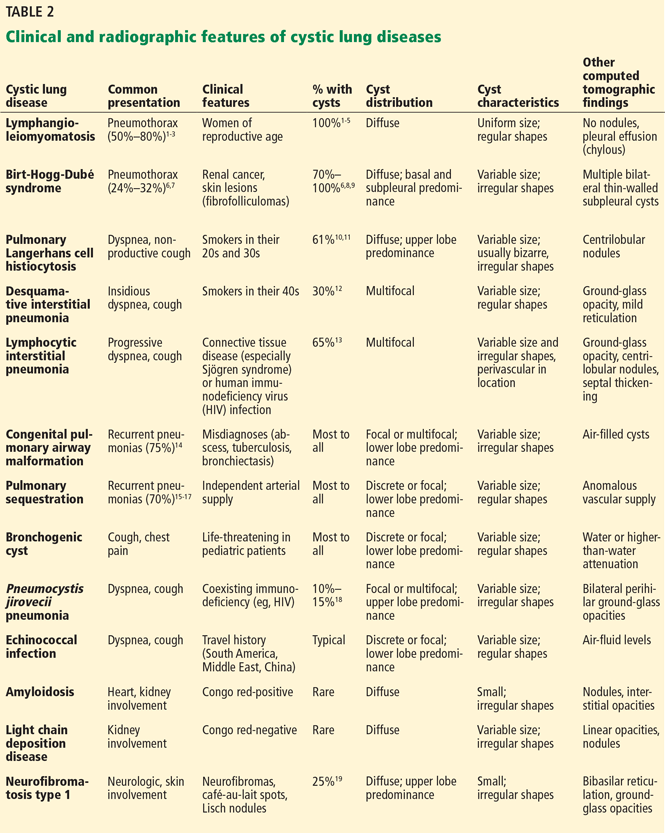
In view of the features of the different types of cystic lung disease, adults with cystic lung disease can be grouped according to their typical clinical presentations (Table 2):
- Insidious dyspnea or spontaneous pneumothorax
- Incidentally found cysts or recurrent pneumonia
- Signs and symptoms of primary pulmonary infection
- Signs and symptoms that are primarily nonpulmonary.
Insidious dyspnea or spontaneous pneumothorax
Insidious dyspnea or spontaneous pneumothorax can be manifestations of lymphangioleiomyomatosis, Birt-Hogg-Dubé syndrome, pulmonary Langerhans cell histiocytosis, desquamative interstitial pneumonia, or lymphocytic interstitial pneumonia.
Lymphangioleiomyomatosis is characterized by abnormal cellular proliferation within the lung, kidney, lymphatic system, or any combination.5 The peak prevalence is in the third to fourth decades of life, and most patients are women of childbearing age.6 In addition to progressive dyspnea on exertion and pneumothorax, other signs and symptoms include hemoptysis, nonproductive cough, chylous pleural effusion, and ascites.7,8
Birt-Hogg-Dubé syndrome is caused by germline mutations in the folliculin (FLCN) gene.9 It is characterized by skin fibrofolliculomas, pulmonary cysts, spontaneous pneumothorax, and renal cancer.10
Pulmonary Langerhans cell histiocytosis is part of the spectrum of Langerhans cell histiocytosis that, in addition to the lungs, can also involve the bone, pituitary gland, thyroid, skin, lymph nodes, and liver.11 It occurs almost exclusively in smokers, affecting individuals in their 20s and 30s, with no gender predilection.12,13 In addition to nonproductive cough and dyspnea, patients can also present with fever, anorexia, and weight loss,13 but approximately 25% of patients are asymptomatic.14
Desquamative interstitial pneumonia is an idiopathic interstitial pneumonia that, like pulmonary Langerhans cell histiocytosis, is seen almost exclusively in current or former smokers, who account for about 90% of patients with this disease. It affects almost twice as many men as women.15,16 The mean age at onset is 42 to 46.15,16 In addition to insidious cough and dyspnea, digital clubbing develops in 26% to 40% of patients.16,17
Lymphocytic interstitial pneumonia is another rare idiopathic pneumonia, usually associated with connective tissue disease, Sjögren syndrome, immunodeficiencies, and viral infections.18–21 It is more common in women, presenting between the 4th and 7th decades of life, with a mean age at diagnosis of 50 to 56.18,22 In addition to progressive dyspnea and cough, other symptoms include weight loss, pleuritic pain, arthralgias, fatigue, night sweats, and fever.23
In summary, in this clinical group, lymphangioleiomyomatosis and Birt-Hogg-Dubé syndrome should be considered when patients present with spontaneous pneumothorax; those with Birt-Hogg-Dubé syndrome also present with skin lesions or renal cancer. In patients with progressive dyspnea and cough, lymphocytic interstitial pneumonia should be considered in those with a known history of connective tissue disease or immunodeficiency. Pulmonary Langerhans cell histiocytosis typically presents at a younger age (20 to 30 years old) than desquamative interstitial pneumonia (smokers in their 40s). Making the distinction, however, will likely require imaging with computed tomography.
Incidentally found cysts or recurrent pneumonia
Incidentally found cysts or recurrent pneumonia can be manifestations of congenital pulmonary airway malformation, pulmonary sequestration, or bronchogenic cyst.
Congenital pulmonary airway malformation, of which there are five types, is the most common pulmonary congenital abnormality. It accounts for up to 95% of cases of congenital cystic lung disease.24,25 About 85% of cases are detected in the prenatal or perinatal periods.26 Late-onset congenital pulmonary airway malformation (arising in childhood to adulthood) presents with recurrent pneumonia in about 75% of cases and can be misdiagnosed as lung abscess, pulmonary tuberculosis, or bronchiectasis.27
Pulmonary sequestration, the second most common pulmonary congenital abnormality, is characterized by a portion of lung that does not connect to the tracheobronchial tree and has its own systemic arterial supply.24 Intralobar sequestration, which shares the pleural investment with normal lung, accounts for about 80% of cases of pulmonary sequestration.28–30 In addition to signs or symptoms of pulmonary infection, patients with pulmonary sequestration can remain asymp-
tomatic (about 25% of cases), or can present with hemoptysis or hemothorax.28–30 In adults, the typical age at presentation is between 20 and 25.29,30
Bronchogenic cyst is usually life-threatening in children. In adults, it commonly causes cough and chest pain.31 Hemoptysis, dysphagia, hoarseness, and diaphragmatic paralysis can also occur.32,33 The mean age at diagnosis in adults is 35 to 40.31,32
In summary, most cases of recurrent pneumonia with cysts are due to congenital pulmonary airway malformation. Pulmonary sequestration is the second most common cause of cystic lung disease in this group. Bronchogenic cyst is usually fatal in fetal development; smaller cysts can go unnoticed during the earlier years and are later found incidentally as imaging abnormalities in adults.
Signs and symptoms of primary pulmonary infections
Signs and symptoms of primary pulmonary infections can be due to Pneumocystis jirovecii pneumonia or echinococcal infections.
P jirovecii pneumonia commonly develops in patients with human immunodeficiency virus infection and low CD4 counts, recipients of hematologic or solid-organ transplants, and those receiving immunosuppressive therapy (eg, glucocorticoids or chemotherapy).
Echinococcal infections (with Echinococcus granulosus or multilocularis species) are more common in less-developed countries such as those in South America or the Middle East, in China, or in patients who have traveled to endemic areas.34
In summary, cystic lung disease in patients with primary pulmonary infections can be diagnosed by the patient’s clinical history and risk factors for infections. Those with human immunodeficiency virus infection and other causes of immunodeficiency are predisposed to P jirovecii pneumonia. Echinococcal infections occur in those with a history of travel to an endemic area.