Safety issues in vasculitis: Infections and immunizations in the immunosuppressed host





ABSTRACTInfectious diseases are a significant cause of morbidity and mortality in immunosuppressed patients, including those with connective tissue diseases. Both disease and treatment contribute to a predisposition to infection in immunocompromised patients. Significant infection and morbidity occur in 25% to 50% of these patients with a median mortality of 5.2% due to common bacterial infections, such as pneumonia or bacteremia, and opportunistic fungal infections such as Pneumocystis. The lungs, skin, urinary tract, blood, and central nervous system are commonly affected. Pathogens such as Pneumocystis jirovecii, Histoplasma capsulatum, Aspergillus species, herpes zoster, JC virus, Nocardia asteroides, and Nocardia species are increasingly prevalent in immunocompromised patients. Improved recognition, diagnosis, and prevention of these infections are needed to enhance outcomes in these patients.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
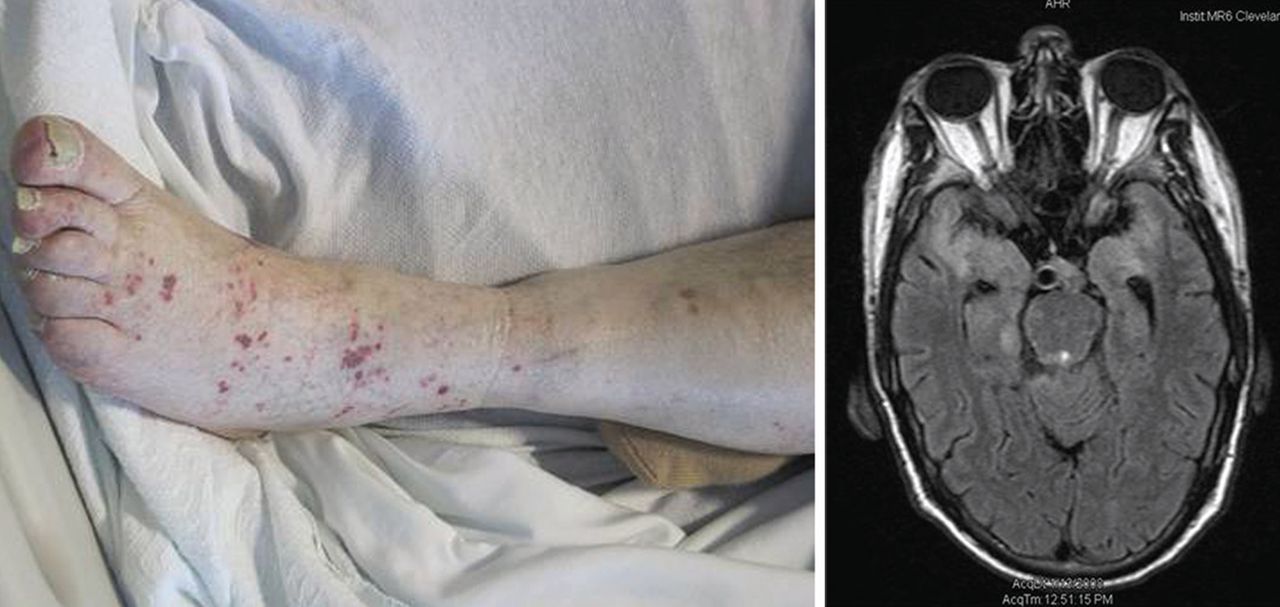
Varicella zoster
JC virus
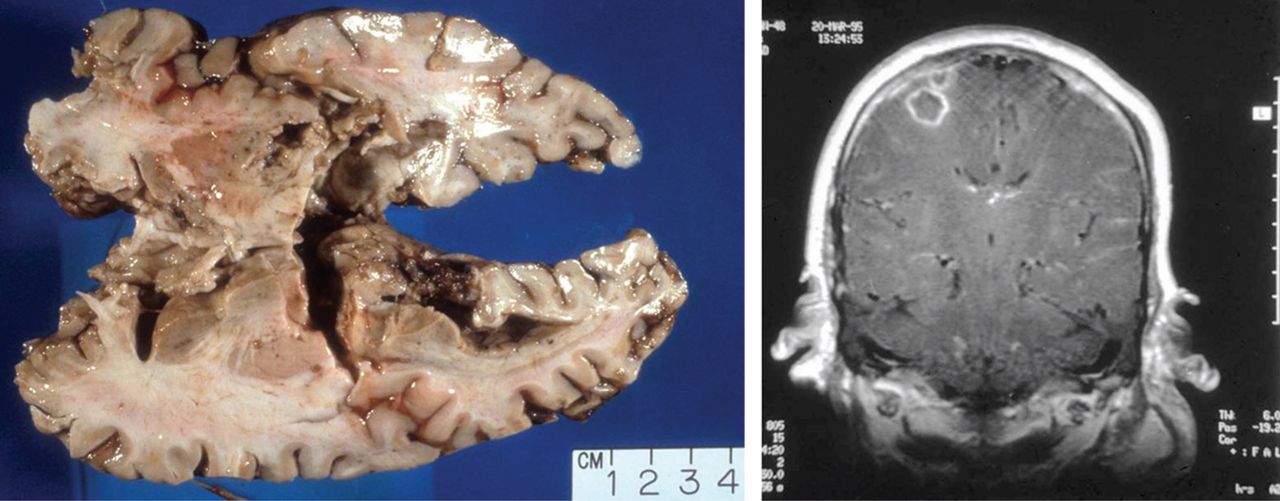
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
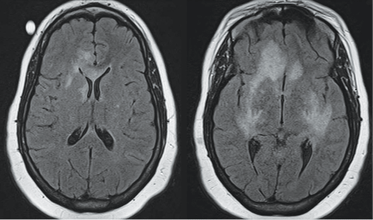
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
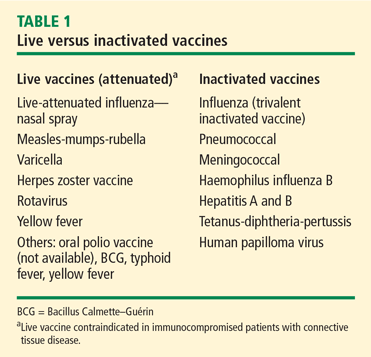
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
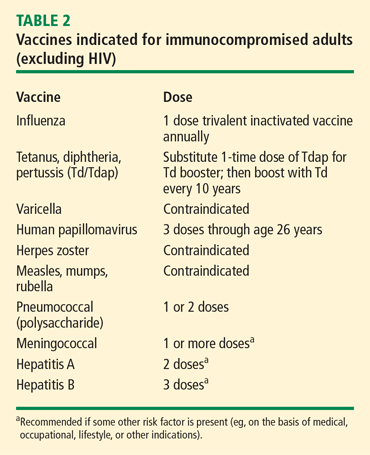
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.