History and current concepts in the pathogenesis of PML





ABSTRACTThe JC virus (JCV), first described in 1971, is responsible for initiation of progressive multifocal leukoencephalopathy (PML), a disease characterized by demyelinating plaques and a classic triad of symptoms consisting of cognitive impairment, visual deficits, and motor dysfunction. To establish a diagnosis of PML, evidence of the presence of JCV DNA in pathologic tissue is necessary. The host range for productive infection of JCV is controlled by factors in the cell nucleus that bind to the viral promoter, initiating transcription of mRNA for the coordinated synthesis of viral proteins. Oligodendrocytes, astrocytes, and CD34+ and CD19+ cells of the immune system have the necessary binding proteins in sufficient concentration to allow lytic infection to occur. A strong link between JCV infection in cells of the immune system and cells of the nervous system points to the importance of the tissue origin of JCV latency, the bone marrow that harbors CD34+ cells. The emergence of PML in patients treated with natalizumab and other immune-altering agents supports this observation and provides new insights into the pathogenic mechanisms of JCV infection.
The neuropathology of progressive multifocal leukoencephalopathy (PML) was first reported in 1958 following examination of brain tissue from two cases of chronic lymphocytic leukemia and one case of Hodgkin lymphoma.1 The classic triad of symptoms of PML—cognitive impairment, visual deficits, and motor dysfunction—had been observed previously but had not been formally described.2
Until PML was discovered in patients with autoimmune diseases treated with biologic therapies that do not directly suppress immunity, PML had been considered a very rare, virus-induced demyelinating disease of the white matter that occurred in immune-compromised patients. The incidence of PML rose sharply in the mid-1980s with the pandemic of human immunodeficiency virus (HIV)-1 infection and continues as an acquired immunodeficiency syndrome–defining illness at a rate of approximately 1% to 3% of HIV-1 seropositive individuals; more recently, it has been seen in approximately 1 in 850 natalizumab-treated individuals who have multiple sclerosis (MS). The incidence of PML in natalizumab-treated MS patients increases with dosing; among those who receive 24 or more doses, the incidence is 1 in 400.
The cause of PML was unknown until 1971, when viral particles were observed by electron microscopy in PML brain lesions and subsequently isolated at the University of Wisconsin, Madison, in cultures of human fetal brain tissue.3 The designation of JC virus (JCV) was derived from the initials of the patient whose brain tissue was used for culture and isolation. Variants in the noncoding region of the genome were then serially identified as Mad 1, Mad 2, and so on, representing the geographic location, Madison, Wisconsin, where the virus was identified.
The JCV, a polyomavirus, is a nonenveloped DNA virus with icosahedral structure containing double-stranded DNA genomes. The circular genome of JCV contains early and late transcription units, the latter of which encodes three virion structural proteins—VPl, VP2, and VP3. Humans generate antibodies directed against the amino terminal end of VP1 and perhaps VP2 and VP3.
JC VIRUS PATHOGENESIS
JCV pathogenesis is studied in cell cultures derived from human fetal brain tissue. In vitro, JCV robustly infects astrocytes, making it important to identify the culture’s cellular phenotypes. A cell line was developed that allows multiplication of JCV and, more recently, human multipotential progenitor cells were isolated and are being grown from the human developing brain at various gestational stages. The lineage pathways of these cells can be differentiated into astrocytes, oligodendrocytes, and neurons. Initiating infection in progenitor cells with JC virions made it possible to determine which cells were susceptible to infection. JCV susceptibility is evident in progenitor-derived astrocytes and glial cells, which reflects the pathologic process in PML brain tissue. Neuronal cells, by contrast, are not susceptible to infection.4
JC VIRUS CHARACTERISTICS: GLOBAL DISTRIBUTION, TRIAD OF SYMPTOMS
Subcortical multifocal white matter lesions are the classic feature of PML on neuroimaging. Seroepidemiology of JCV has revealed ubiquitous distribution, with 50% to 60% of adults aged 20 to 50 years demonstrating antibody to JCV.5 The percentage of the population with antibody increases with age, but may vary among geographic regions. Prevalence is lower among remote populations.
Although the initial site of JCV infection is not well characterized, we know that the primary infection is not in the brain. The JCV has a selective tropism for replication in glial cells in the human brain, but the absence of an animal model for PML has hindered our understanding of the JCV migration to the brain and the initiation and development of central nervous system infection.
Although humans carry JCV-specific antibodies, the clinical significance of these antibodies is unknown. Antibody levels rise during active infection, at times to very high titers, but offer no protection. T-cell–mediated immune responses directed to structural and nonstructural proteins are important in controlling infection.
A high index of suspicion for PML is warranted in individuals who demonstrate the classic triad of symptoms (cognitive impairment, visual deficits, and motor dysfunction) and in whom magnetic resonance imaging shows evidence of demyelinated plaque lesions; however, evidence of the presence of JCV DNA in pathologic tissue is necessary to confirm a diagnosis of PML.
The development of an in situ DNA hybridization assay using a biotinylated probe has facilitated identification of JCV DNA in the infected nuclei of the pathologic tissue. The presence of JCV DNA in cerebrospinal fluid (CSF) samples can be detected using a quantitative polymerase chain reaction assay, targeting the viral genome in the amino terminal end of the viral T protein.6 This T protein coding region was targeted because it does not crossreact, even with other human polyomaviruses, and it is intolerant of mutations. This assay is certified by the Clinical Laboratory Improvement Amendments, licensed by the National Institutes of Health; it is the most sensitive (to levels of 10 copies/mL sample) assay available.
JC VIRUS SUSCEPTIBILITY FACTORS
Despite the high prevalence of JCV infection, PML is rare, suggesting important barriers to its development. Although the receptor for JCV has been identified as alpha 2,6-linked sialic acid, the host range for productive infection is controlled by factors within the cell nucleus that bind to the viral promoter; this process initiates transcription of mRNA for the coordinated synthesis of viral proteins. Only certain cells have the necessary DNA binding proteins in high enough concentrations to allow lytic infection to take place, spreading by cell-to-cell contact. These cells include oligodendrocytes, the primary target for JCV, whose destruction leads to PML; astrocytes; and the CD34+ and CD19+ cells of the immune system. JCV can also be found in urine, at times in very high concentrations. It is present in the uroepithelial cells and multiplies without apparent pathologic consequences. Virus isolated from the urine has not been grown in cell culture systems in the laboratory setting.
Bone marrow CD34+ hematopoietic progenitor cells represent a potential pathway of JCV pathogenesis: in six people with PML, latent JCV DNA was demonstrated in pathologic tissue from lymph, spleen, or bone marrow biopsies taken months to years before the patient developed neurologic disease.7
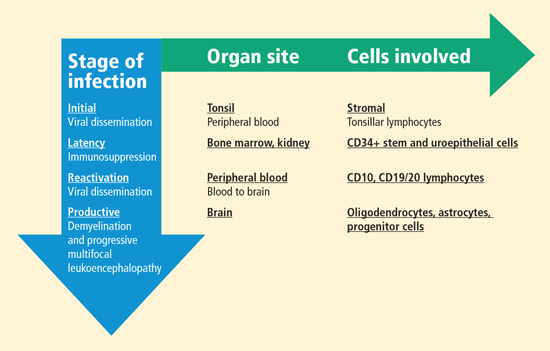
Upon immunosuppression, reactivation of the virus occurs, with evidence of the virus found in CD10 and CD19/20 lymphocytes in the peripheral blood of some individuals. Blood-to-brain viral dissemination results in infection of oligodendrocytes, astrocytes, and progenitor cells.