Neuroscience and heart-brain medicine: The year in review





ABSTRACT
Important recent publications in the area of neuroscience and heart-brain medicine center largely around three topics: (1) mechanisms of cardiac sympathetic denervation in Parkinson disease, (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone. Findings by Orimo et al support a centripetal, retrograde pathogenetic process involving alpha-synuclein deposition and degeneration of cardiac noradrenergic neurons in Parkinson disease. Several studies suggest that processes increasing cytoplasmic monoamines lead to neuronal loss from auto-oxidation or enzymatic oxidation. Lack of correlation between commonly used indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover casts doubt on the validity of power spectral analysis to indicate cardiac sympathetic tone.
CYTOPLASMIC MONOAMINE METABOLITES AS AUTOTOXINS
Current concepts about mechanisms of PD emphasize pathologic alpha-synuclein accumulation, oxidative injury, impaired proteasomal or mitochondrial functions, neuroinflammation, or abnormal kinase signaling. These concepts do not explain relatively selective nigrostriatal dopaminergic and cardiac noradrenergic denervation in PD.
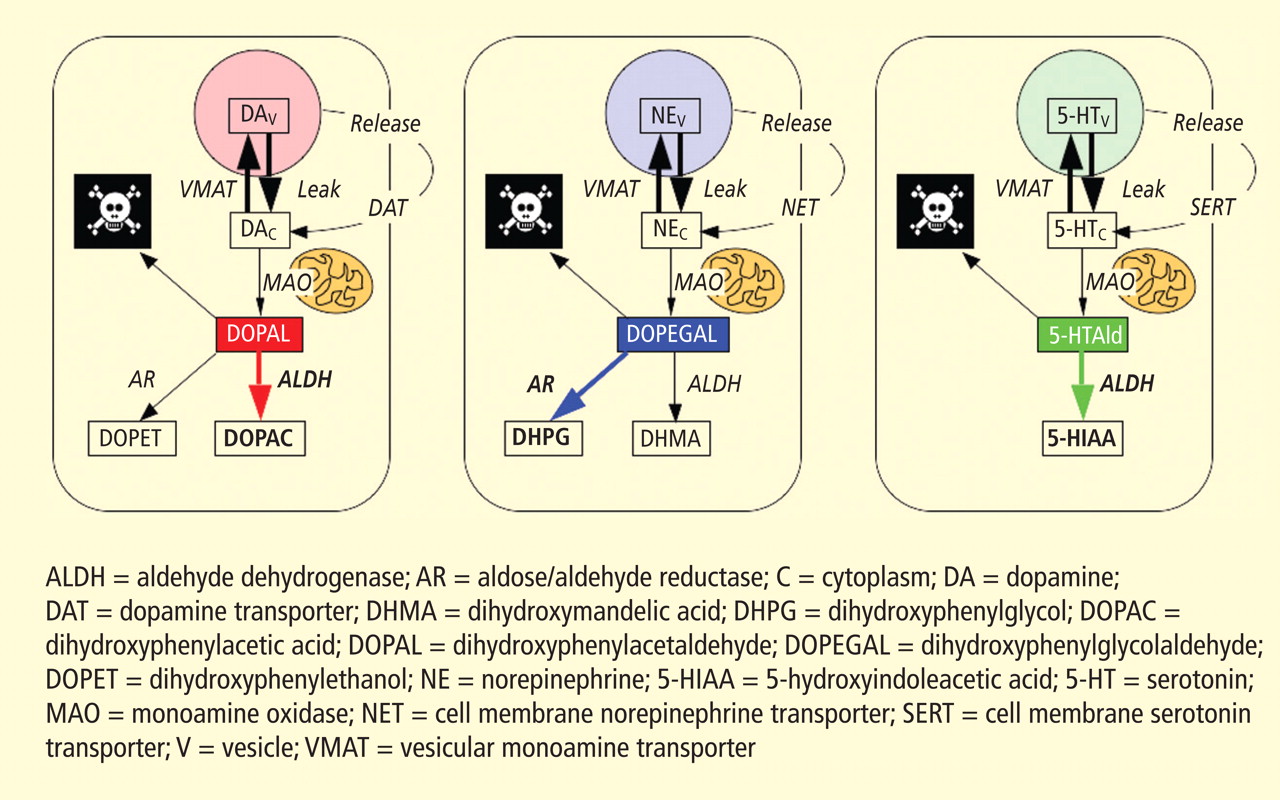
A potential explanation is that cytoplasmic catecholamine metabolites are autotoxins (Figure 3). The mechanisms of autotoxicity include spontaneous auto-oxidation, to form quinones and chromes leading to increased production of reactive oxygen species, and enzymatic oxidation.
Catecholamines in the neuronal cytoplasm undergo enzymatic oxidative deamination to form catecholaldehydes (dihydroxyphenylacetaldehyde [DOPAL] from dopamine), which are cytotoxic, as predicted by Blaschko more than a half century ago.10 DOPAL is detoxified mainly by aldehyde dehydrogenase (ALDH). In the substantia nigra, aldehyde dehydrogenase 1A1 (ALDH1A1) is the main isoform of ALDH, and postmortem studies have noted decreased nigral ALDH1A1 gene expression11,12 and protein content13 in PD patients.
All neurons express alpha-synuclein. Current concepts about mechanisms also do not explain the relatively selective aggregation of alpha-synuclein in catecholaminergic neurons. Alpha-synuclein appears to play a role in the cycling of catecholamines across vesicular and cell membranes.14
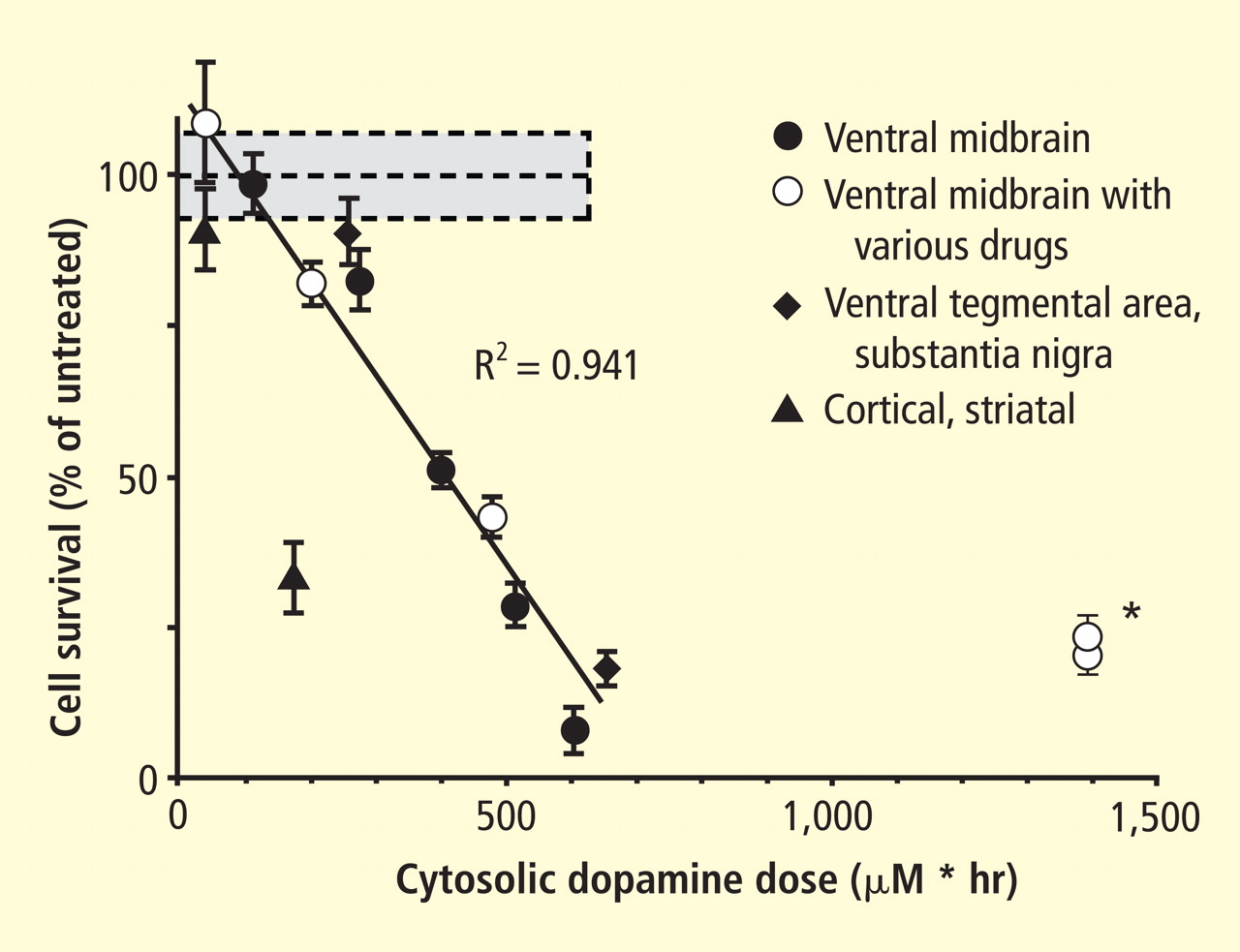
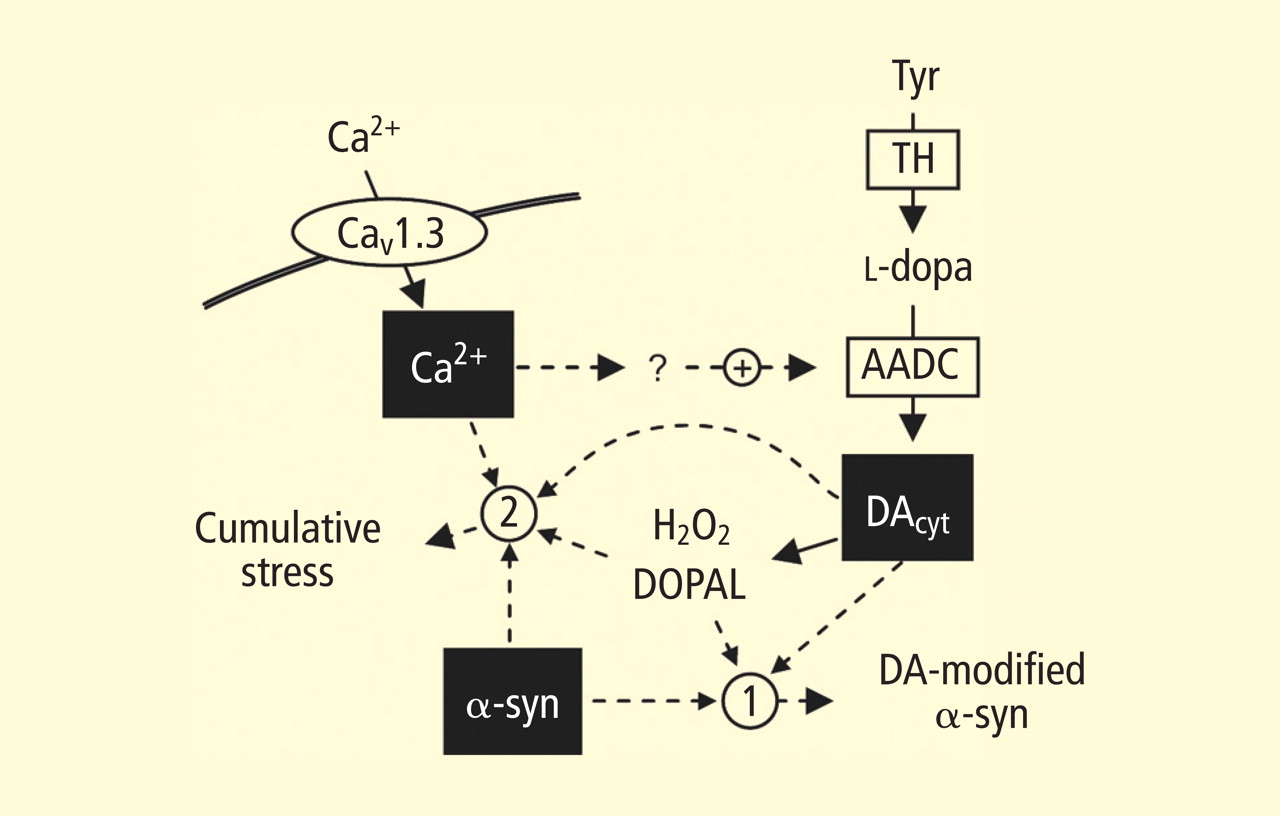
Under resting conditions, most catecholamine turnover results from leakage from vesicular stores into the cytoplasm and subsequent oxidative deamination by monoamine oxidase. Ordinarily, however, catecholamines in the cytoplasm are efficiently recycled back into the vesicles via the type 2 vesicular monoamine transporter (VMAT-2). Accordingly, interference with VMAT functions would be expected to tend to build up cytoplasmic catecholamines, with potentially cytotoxic consequences. In 2007, Caudle et al reported that mice with severely decreased VMAT-2 have aging-associated decreases in striatal dopamine that begin in the terminal fields, alpha-synuclein deposition in substantia nigra neurons, and l-dopa–responsive behavioral deficits.18 More recently the same group noted nonmotor signs associated with PD in VMAT-2–deficient mice, such as anosmia, gastrointestinal hypomotility, sleep disturbances, anxiety, and depression.19 Since VMAT-2 serves to recycle not only dopamine but also norepinephrine and serotonin, this single abnormality could help explain loss of all three types of monoaminergic neurons in PD.
Finally, Pena-Silva et al recently tested whether serotonin induces oxidative stress in human heart valves.20 They showed that in heart valves from explanted human hearts not used for transplantation, incubation of homogenates of cardiac valves and blood vessels with serotonin increased generation of the superoxide free radical. Inhibitors of monoamine oxidase prevented this effect. Dopamine also increased superoxide levels in heart valves, and this effect was also attenuated by monoamine oxidase inhibition. These findings fit with the concept that the aldehydes produced by the action of monoamine oxidase on cytoplasmic monoamines generate toxic free radicals.