David S. Goldstein, MD, PhD Clinical Neurocardiology Section, Clinical Neurosciences Program, Division of Intramural Research, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD
Correspondence: David S. Goldstein, MD, PhD, Building 10, Room 5N220, 10 Center Drive, MSC-1620, Bethesda, MD 20892–1620; goldsteind@ninds.nih.gov
Dr. Goldstein reported that he has no financial relationships that pose a potential conflict of interest with this article.
Some of the research summarized here was supported by the intramural research program of the National Institutes of Health.
ABSTRACT
Important recent publications in the area of neuroscience and heart-brain medicine center largely around three topics: (1) mechanisms of cardiac sympathetic denervation in Parkinson disease, (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone. Findings by Orimo et al support a centripetal, retrograde pathogenetic process involving alpha-synuclein deposition and degeneration of cardiac noradrenergic neurons in Parkinson disease. Several studies suggest that processes increasing cytoplasmic monoamines lead to neuronal loss from auto-oxidation or enzymatic oxidation. Lack of correlation between commonly used indices from power spectral analysis of heart rate variability and cardiac norepinephrine spillover casts doubt on the validity of power spectral analysis to indicate cardiac sympathetic tone.
This review highlights important recent publications in the area of neuroscience and heart-brain medicine. Abnormalities of regulation of the circulation by catecholamine systems figure as a general theme of the topics highlighted. These topics, which are reviewed in turn below, are (1) mechanisms of cardiac sympathetic denervation in Parkinson disease (PD), (2) cytoplasmic monoamine metabolites as autotoxins, and (3) the validity of power spectral analysis of heart rate variability to indicate cardiac sympathetic tone.
MECHANISMS OF CARDIAC SYMPATHETIC DENERVATION IN PARKINSON DISEASE
The movement disorder component of PD is well recognized as resulting from loss of dopaminergic neurons in the nigrostriatal system of the brain. The finding of low myocardial 6-[18F]fluorodopamine–derived radioactivity by positron emission tomography provided the first neuroimaging evidence for loss of catecholaminergic neurons outside the brain in PD.1 Many reports using 123I-metaiodobenzylguanidine scanning have concurred with this finding. Beginning in the early 2000s, post-mortem neuropathologic studies demonstrated virtually absent immunoreactivity for tyrosine hydroxylase, the rate-limiting enzyme in norepinephrine biosynthesis, in epicardial nerves in PD.2,3 These results provided clues to the mechanism of autonomic dysfunction in PD, a prominent nonmotor manifestation of the disease.
With kind permission from Springer Science+Business Media: Acta Neuropathologica,
Figure 1. Tyrosine hydroxylase immunoreactivity (THir) in epicardial nerve from (A) a control subject and (B) a patient with familial Parkinson disease due to duplication of the gene encoding alpha-synuclein (PARK4).6
Alpha-synuclein is a key protein in the pathogenesis of PD. It is abundant in Lewy bodies and Lewy neurites, and mutations or multiplications of the gene that encodes it cause rare inherited forms of PD. In 2001 we reported evidence for cardiac sympathetic denervation, neurogenic orthostatic hypotension, and baroreflex failure in familial PD from mutation of the gene encoding alpha-synuclein.4 Subsequently we reported analogous denervation in familial PD from triplication of the normal gene.5 This past year Orimo’s group in Tokyo provided the first pathological confirmation of cardiac sympathetic denervation in familial PD from inherited alpha-synucleinopathy, based on severely decreased epicardial neuronal tyrosine hydroxylase immunoreactivity (Figure 1).6 In contrast, patients with familial PD from parkin gene mutation, which is not thought to be a Lewy body disease, have been found to have normal cardiac 123I-metaiodobenzyl-guanidine–derived radioactivity and normal epicardial neuronal tyrosine hydroxylase immunoreactivity.7 These findings establish a link between alpha-synucleinopathy and cardiac sympathetic denervation.
Some individuals who die without clinical parkinsonism have Lewy bodies detected pathologically. Growing evidence shows that incidental Lewy body disease represents early, presymptomatic PD.8 Orimo’s group therefore studied cardiac tissues and paravertebral sympathetic ganglia from patients with incidental Lewy body disease.9 Postmortem tissues were likewise obtained from comparison subjects with multiple system atrophy and from control subjects. Immunohistochemical analyses were performed using antibodies against tyrosine hydroxylase, phosphorylated neurofilament as a marker of axons, and phosphorylated alpha-synuclein as a marker of abnormal alpha-synuclein deposits. Key findings from this study9 were as follows:
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
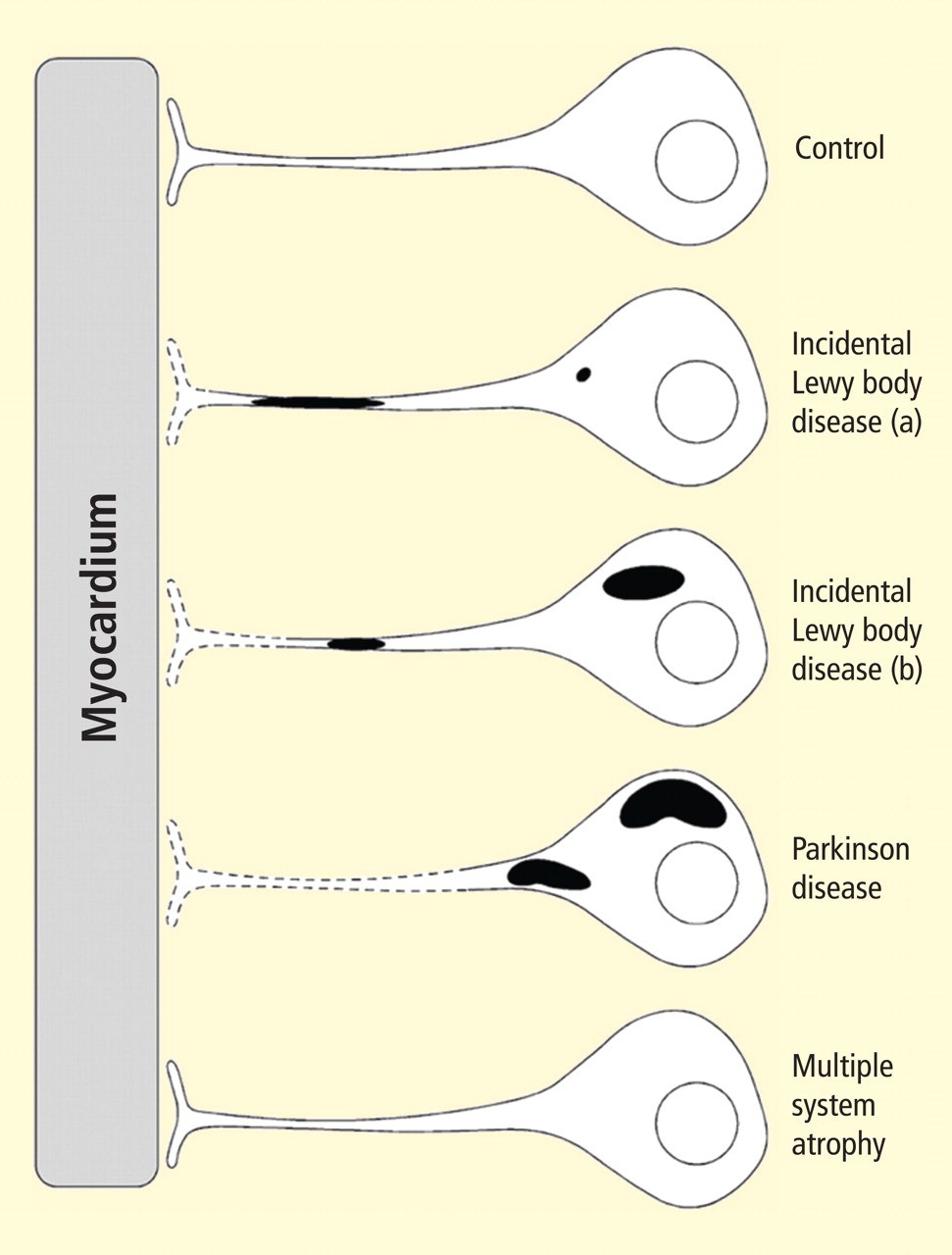
Figure 2. Concept diagram of the pathogenetic sequence of cardiac sympathetic denervation. In incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons (a), alpha-synuclein aggregates (black shading) accumulate abundantly in the distal axons but sparsely in the paravertebral sympathetic ganglia. In contrast, in incidental Lewy body disease with decreased THir axons (b), alpha-synuclein aggregates diminish in the distal axons but increase in the paravertebral sympathetic ganglia. In Parkinson disease, alpha-synuclein aggregates disappear in the distal axons and accumulate much more abundantly in the paravertebral sympathetic ganglia. In multiple system atrophy, alpha-synuclein aggregates are generally not observed (as in controls), with a few exceptions. Dotted lines indicate degeneration of THir axons.9
Alpha-synuclein aggregates in distal epicardial nerve fascicles were more abundant in incidental Lewy body disease with preserved tyrosine hydroxylase–immunoreactive (THir) axons than in incidental Lewy body disease with decreased THir axons (Figure 2).
Alpha-synuclein aggregates in the epicardial nerve fibers were closely related to the disappearance of THir axons.
In incidental Lewy body disease with preserved THir axons, alpha-synuclein aggregates were consistently more abundant in the epicardial nerves than in the paravertebral sympathetic ganglia (Figure 2).
Distally dominant accumulation of alpha-synuclein aggregates was reversed in incidental Lewy body disease with decreased THir axons and in PD, because both conditions involve fewer alpha-synuclein aggregates in axons and more abundant aggregates in the paravertebral sympathetic ganglia (Figure 2).
Thus, accumulation of alpha-synuclein aggregates in distal cardiac sympathetic axons precedes aggregation in neuronal somata or ganglionic neurites, heralding centripetal degeneration of cardiac sympathetic nerves in PD. This chronological and dynamic relationship between alpha-synuclein aggregation and distally dominant degeneration of cardiac noradrenergic nerves may represent the pathological mechanism behind a common degenerative process in PD.
In conclusion, cardiac noradrenergic denervation in Lewy body diseases, even in early stages, accounts for reduced cardiac uptake of 123I-metaiodobenzylguanidine and 6-[18F]fluorodopamine in PD. Alpha-synuclein aggregation appears to be intimately involved in the cardiac noradrenergic denervation that attends Lewy body diseases. The pathogenetic process seems to proceed in a centripetal, retrograde direction.






 Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.
Reprinted from Brain (Orimo S, et al. Axonal α-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 2008; 131:642–650) by permission of Oxford University Press.