When ‘blue babies’ grow up: What you need to know about tetralogy of Fallot





ABSTRACTMost babies born with tetralogy of Fallot undergo corrective surgery and survive to adulthood. However, as they get older they are prone to a number of long-term problems, and they often do not receive expert-level follow-up care. This review of the adult complications of tetralogy of Fallot should help primary care practitioners identify these patients, make appropriate and timely referrals, and educate patients and their families.
KEY POINTS
- The major long-term complication of tetralogy of Fallot repair is pulmonary valve insufficiency, which leads to right heart failure. Other problems include atrial and ventricular arrhythmias and sudden cardiac death.
- Surgical pulmonary valve replacement is the standard of care, but the optimal time to do this is unclear.
- Novel and experimental therapies include percutaneous pulmonary valve replacement and medical therapy with pulmonary arterial vasodilators.
Children born with tetralogy of Fallot and other congenital heart defects are living longer—long enough for new problems to arise, and, eventually, to present to your clinic. In primary care, the presentation of tetralogy of Fallot is still rare, but it is becoming more common.
Congenital heart disease was once solely a pediatric specialty, but adults who have been treated for these conditions now outnumber children with congenital heart conditions.1–4 More than 85% of infants with congenital heart disease are now expected to reach adulthood.5,6 For those with tetralogy of Fallot, the most common form of cyanotic congenital heart disease, the 40-year survival rate is now at least 90%.5
But these former “blue babies” eventually have serious problems. Most develop pulmonary valve insufficiency (regurgitation), which, over time, can result in right ventricular volume overload, enlargement, and dysfunction. 7–10 These problems lead to arrhythmias, the most significant cause of illness and death in these patients.11–13 Ventricular and atrial arrhythmias occur in up to 35% of patients with tetralogy of Fallot, and over a follow-up period of up to 30 years the incidence of sudden cardiac death is 6%.14
Furthermore, because many patients have no symptoms in early adulthood, they are often lost to follow-up, potentially missing the opportunity to have complications treated before they become irreversible. Recent data suggest that most patients who present with symptoms had stopped seeing a cardiologist about 10 years before.15
The challenge for primary care clinicians is to identify these patients in their practice, to recognize the early signs and symptoms of a worsening condition, and to refer and treat before cardiac damage becomes irreversible.
ONE IN 3,600 LIVE BIRTHS
Tetralogy of Fallot occurs in approximately 1 in 3,600 live births or 3.5% of infants born with congenital heart disease.6 It is the most common type of cyanotic congenital heart disease, accounting for 10% of all cases.16 Patients in whom it has been repaired are the biggest group of adults with complex congenital heart disease. At Cleveland Clinic, it is the reason for 23% of new referrals to our adult congenital cardiology clinic, second only to atrial septal defects (33%).
FOUR DISTINCT FEATURES
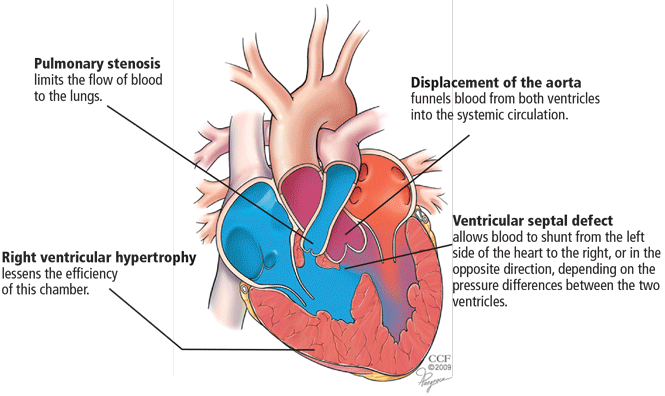
- Pulmonary stenosis (subvalvar, valvar, or both subvalvar and valvar)
- Ventricular septal defect
- Hypertrophy of the right ventricle
- Rightward deviation of the aortic valve, so that it overrides the ventricular septum; this can range from minimal overriding of the aorta and trivial pulmonary stenosis to up to 90% override and frank pulmonary atresia.
The aorta, receiving blood from both ventricles, is usually dilated. It arises from a right-sided arch in about 25% of patients and may override the septum so much that more than 50% of the blood flow comes from the right ventricle.17 In such cases, whether the patient has true tetralogy of Fallot or a double-outlet right ventricle with pulmonic stenosis may be ambiguous. Though controversial, the latter condition is generally distinguished by a ventricular septal defect that is integral to the left ventricular outflow tract and by lack of fibrous continuity between the aortic and mitral valves.18
SURGERY HAS EVOLVED
Surgical repair has been performed since the 1950s, and the perioperative death rate has fallen to less than 1% at most experienced centers.19
In the past, surgeons often placed a shunt between a systemic artery and the pulmonary artery as a palliative measure to improve oxygenation in infants with tetralogy of Fallot, waiting until the child was older to remove the shunt and repair the defects definitively.
Now, however, they generally favor repairing the heart in the initial procedure. This involves patching the ventricular septal defect, widening the infundibulum, and repairing the pulmonary valve or patching the annulus. Transannular patching opens the entire right ventricular outflow tract, but it crosses the pulmonary valve, and this is what eventually results in severe pulmonary insufficiency and its complications.20 For this reason, surgeons at most institutions now favor valve-sparing procedures rather than transannular patching, whenever possible.