Clinical manifestations of hyperuricemia and gout





ABSTRACT
Biologically significant hyperuricemia occurs when serum urate levels exceed urate solubility, ie, at approximately 6.8 mg/dL. At serum urate levels above this threshold, manifestations of chronic crystal deposition, including gouty arthritis, may occur, although asymptomatic hyperuricemia often persists for many years without progression. Intercritical asymptomatic periods follow the resolution of acute gout flares, but crystals remain in the joint during these intervals and further deposition may continue silently. Ultimately this may lead to persistent attacks, chronic pain, and, in some patients, joint damage.
KEY POINTS
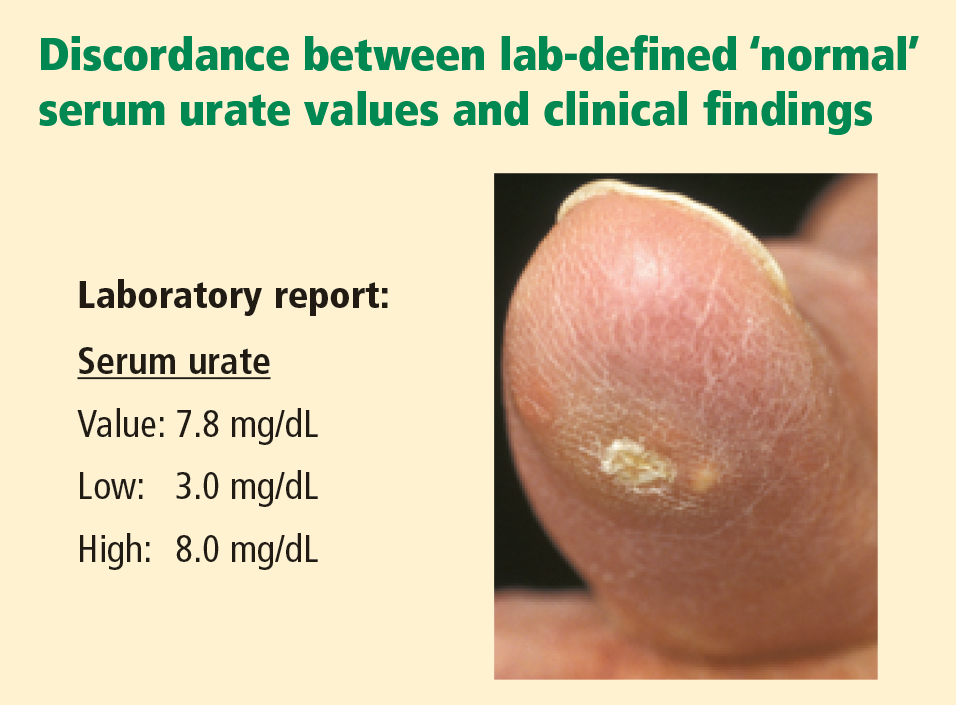
- Clinically significant hyperuricemia includes serum urate levels that fall within the population-defined “normal” range of many clinical laboratories.
- There is no reliable way to predict the likelihood that gout will develop in a given hyperuricemic patient. Treatment of asymptomatic hyperuricemia is not generally recommended.
- Untreated, an initial acute gout attack resolves within 3 to 14 days. Subsequent attacks tend to last longer and may involve more joints.
- Chronic gout can mimic rheumatoid or psoriatic arthritis.
Uric acid—urate in most physiologic fluids—is an end product of purine degradation. The serum urate level in a given patient is determined by the amount of purines synthesized and ingested, the amount of urate produced from purines, and the amount of uric acid excreted by the kidney (and, to a lesser degree, from the gastrointestinal tract).1 A major source of circulating urate is the metabolized endogenous purine. Renal excretion is likely determined by genetic factors that dictate expression of uric acid transporters, as well as by the presence of organic acids, certain drugs, hormones, and the glomerular filtration rate. A small minority of patients will have increased production of urate as a result of enzymopathies, chronic hemolysis, or rapidly dividing tumors, psoriasis, or other disorders characterized by increased turnover of cells.
Humans do not have a functional enzyme (uricase) to break down urate into allantoin, which is more soluble and readily excreted. There may have been genetic pressures that explain why functional uricase was lost and why humans have relatively high urate levels compared with other species.2 If higher levels of serum urate are clinically detrimental, one would think that humans could have evolved an efficient way to excrete it. Instead, we excrete uric acid inefficiently as a result of active reabsorption in the proximal renal tubule. We have higher levels of serum urate than most other species, and we are predisposed to develop gouty arthritis and perhaps other sequelae of hyperuricemia, including hypertension, the metabolic syndrome, and coronary artery disease.
CLINICALLY SIGNIFICANT HYPERURICEMIA VS LAB-DEFINED HYPERURICEMIA
Clinically significant hyperuricemia is a serum urate level greater than 6.8 mg/dL, although the population-defined “normal” urate level indicated by the clinical laboratory is higher. At levels above 6.8 mg/dL, urate exceeds its solubility in most biological fluids.
SERUM URATE CAN VARY BY SEX, AGE, DIET
Men generally have higher serum urate levels than premenopausal women; serum urate levels increase in women after menopause. For years these findings were attributed to an estrogen effect, but the mechanism was not well understood. Recently a specific transporter (urate transporter 1 [URAT1]) has been identified in the proximal tubule of the kidney3 that seems primarily responsible for the reabsorption of uric acid. Estrogen has a direct effect on the expression of this transporter. It also seems that the hypouricemic effects of probenecid and losartan, as well as the hyperuricemic effects of organic acids and high insulin levels, may be mediated via modulation of URAT1 activity.
Urate values tend to be lower in children, and urate levels are generally affected only modestly by diet.4 Epidemiologic studies, however, have linked increased ingestion of red meats and low ingestion of dairy foods with an increased incidence of gout.5 Acute alcohol ingestion can cause fluctuations in the serum urate levels and may precipitate acute gout attacks.
HYPERURICEMIA LEADING TO GOUT
Urate concentrations greater than 6.8 mg/dL may result in the deposition of urate crystals in the tissues around joints and in other soft tissue structures (tophi). Why this occurs in only some patients is not known. Crystals, when mobilized from these deposits, can provoke the acute gouty flare. The tophi are not usually hot or tender. Biopsy of a tophus reveals a chronic granulomatous inflammatory response around the sequestered crystals. However, the tophi are not inert; the uric acid can be mobilized by mass action effect if the urate in surrounding fluid is reduced. If tophi are adjacent to bone, erosion into bone may occur.
CLINICAL PROGRESSION OF HYPERURICEMIA AND GOUT: FOUR STAGES OF A CHRONIC DISEASE
Although there is significant heterogeneity in the expression of gout, we can conceptualize a prototypic progression from asymptomatic hyperuricemia to chronic gouty arthritis.
Stage 1: Asymptomatic hyperuricemia. At a serum urate concentration greater than 6.8 mg/dL, urate crystals may start to deposit. During this period of asymptomatic hyperuricemia, urate deposits may directly contribute to organ damage. This does not occur in everyone, however, and at present there is no evidence that treatment is warranted for asymptomatic hyperuricemia.
Stages 2 and 3: Acute gout and intercritical periods. If sufficient urate deposits develop around joints, and if the local milieu or some trauma triggers the release of crystals into the joint space, a patient will suffer acute attacks of gout. These flares are self-resolving but are likely to recur. The intervals between attacks are termed “intercritical periods.” During these periods, crystals may still be present at a low level in the fluid, and are certainly present in the periarticular and synovial tissue, providing a nidus for future attacks.
Stage 4: Advanced gout. If crystal deposits continue to accumulate, patients may develop chronically stiff and swollen joints. This advanced stage of gout is relatively uncommon but is avoidable with therapy.
Progression is variable
The progression from asymptomatic hyperuricemia to advanced gout is quite variable from person to person. In most people it takes many years to progress, if it does so at all. In patients treated with cyclosporine following an organ transplant, the progression can be accelerated, although the reasons are not fully understood.
ASYMPTOMATIC HYPERURICEMIA: TO TREAT OR NOT TO TREAT?
Clues for predicting the likelihood that an individual patient with asymptomatic hyperuricemia will develop articular gout are elusive. Campion and colleagues presented data on men without a history of gout who were grouped by serum urate level and followed over a 5-year period.6 The higher the patient’s urate level, the more likely that he would have a gouty attack during the 5 years. In this relatively young population of hyperuricemic men (average age of 42 years), less than 30% developed gout over this short period.
The dilemma is how to predict who is most likely to get gout and will benefit from early urate-lowering treatment, and who will not. Currently, clinicians have no reliable way of predicting the likelihood of gout development in a given hyperuricemic patient. A history of organ transplantation, the continued need for diuretics, an extremely high urate level, alcohol ingestion, low dietary consumption of dairy products, high consumption of meat and seafood, and a family history of gout at a young age suggest a higher risk of gouty arthritis. At present, treatment of asymptomatic hyperuricemia in order to prevent gouty arthritis is not generally recommended.