Soft-tissue sarcomas: Overview of management, with a focus on surgical treatment considerations





ABSTRACT
Patients with soft-tissue sarcomas generally present with a mass that is increasing in size; the presence or absence of pain is not typically predictive of malignancy. While magnetic resonance imaging (MRI) can identify a few soft-tissue lesion types as benign, diagnosis of most lesions requires a careful biopsy, preferably performed by or in consultation with the surgeon who would do an eventual resection. If biopsy confirms a diagnosis of sarcoma, MRI-guided surgical resection with a wide margin is the mainstay of treatment. Neoadjuvant radiation therapy and chemotherapy have not been of proven benefit in well-controlled studies but are frequently used as adjuncts. Resections with wide margins are generally associated with a low (< 10%) risk of recurrence.
Soft-tissue sarcomas are tumors of the mesenchymal system, and half develop in the extremities.1 Although patients with soft-tissue sarcomas have been treated with a combination of surgery, radiation therapy, and chemotherapy, it remains unclear whether either radiation or chemotherapy improves outcomes for these patients. Soft-tissue sarcomas are therefore currently treated with surgical resection when possible, with or without chemotherapy or radiation.
Even though multimodal therapy for patients with these tumors is controversial, a multidisciplinary conference among the many providers who may be involved in the management these patients—orthopedic, medical, and radiation oncologists, as well as the referring primary care physician, plastic and reconstructive surgeons, physical therapists, and radiologists and pathologists with expertise in these tumors—is helpful.2 This article presents an overview of the management of these patients, with a focus on the mainstay treatment, surgical resection. The roles of chemotherapy and radiation therapy for soft-tissue sarcomas, while touched upon here, are detailed in the final two articles in this supplement.
HISTOLOGIC GRADING AND THERAPY IMPLICATIONS
The prognosis of soft-tissue sarcomas correlates with histopathologic grade, and a three-grade system appears to be more accurate than a two-grade system.3 In general, low-grade lesions (grade 1) are unlikely to metastasize and are therefore less likely to need treatment with chemotherapy or radiation, as the risks of these therapies would most likely outweigh any benefit in terms of local control.
Specifically, the risk of radiation involves debilitation of local wound healing and the chance of dedifferentiation of low-grade lesions to higher-grade lesions with more metastatic potential. Grade 2 and 3 lesions are usually considered high-grade and are more likely to be treated with radiation and chemotherapy. Radiation is frequently used in patients with high-grade lesions when anticipated margins or actual margins are less than 1 cm.4–6
Chemotherapy’s lack of proven efficacy for soft-tissue sarcomas likely stems from poor understanding of the pathophysiology, molecular biology, and even some aspects of the natural history of these uncommon and heterogeneous tumors. There are more than 50 subtypes of soft-tissue sarcoma,7,8 and this heterogeneity has likely contributed to the difficulty of identifying chemotherapeutic agents that are highly active against these diseases.9
THE ROLE OF FAMILIAL GENETICS
Developing effective chemotherapeutic strategies may depend on grouping soft-tissue sarcomas more homogeneously. To compare like lesions with like lesions, molecular analysis and even molecular signatures may be of assistance. Along these lines, critical mutations and translocations have been described for several soft-tissue sarcoma subtypes.
Li-Fraumeni syndrome is an autosomal dominant cancer predisposition syndrome caused by germline mutations (ie, in every cell) in the p53 gene.10 Patients with Li-Fraumeni syndrome have an increased risk of developing soft-tissue sarcomas.1,11
Neurofibromatosis type 1 is caused by germline mutations in the NF1 gene, and malignant peripheral nerve sheath tumors occur within neurofibromas in neurofibromatosis patients and typically have additional mutations in CDKN2A or p53.9 INI1 loss is seen in all cases of extrarenal rhabdoid tumors and has been reported in a subset of epithelioid sarcomas (those occurring in proximal/axial regions).9,12 Delineation and greater understanding of these genetic abnormalities may lead to more effective medical therapy.
EVALUATION OF SUSPICIOUS SOFT-TISSUE MASSES
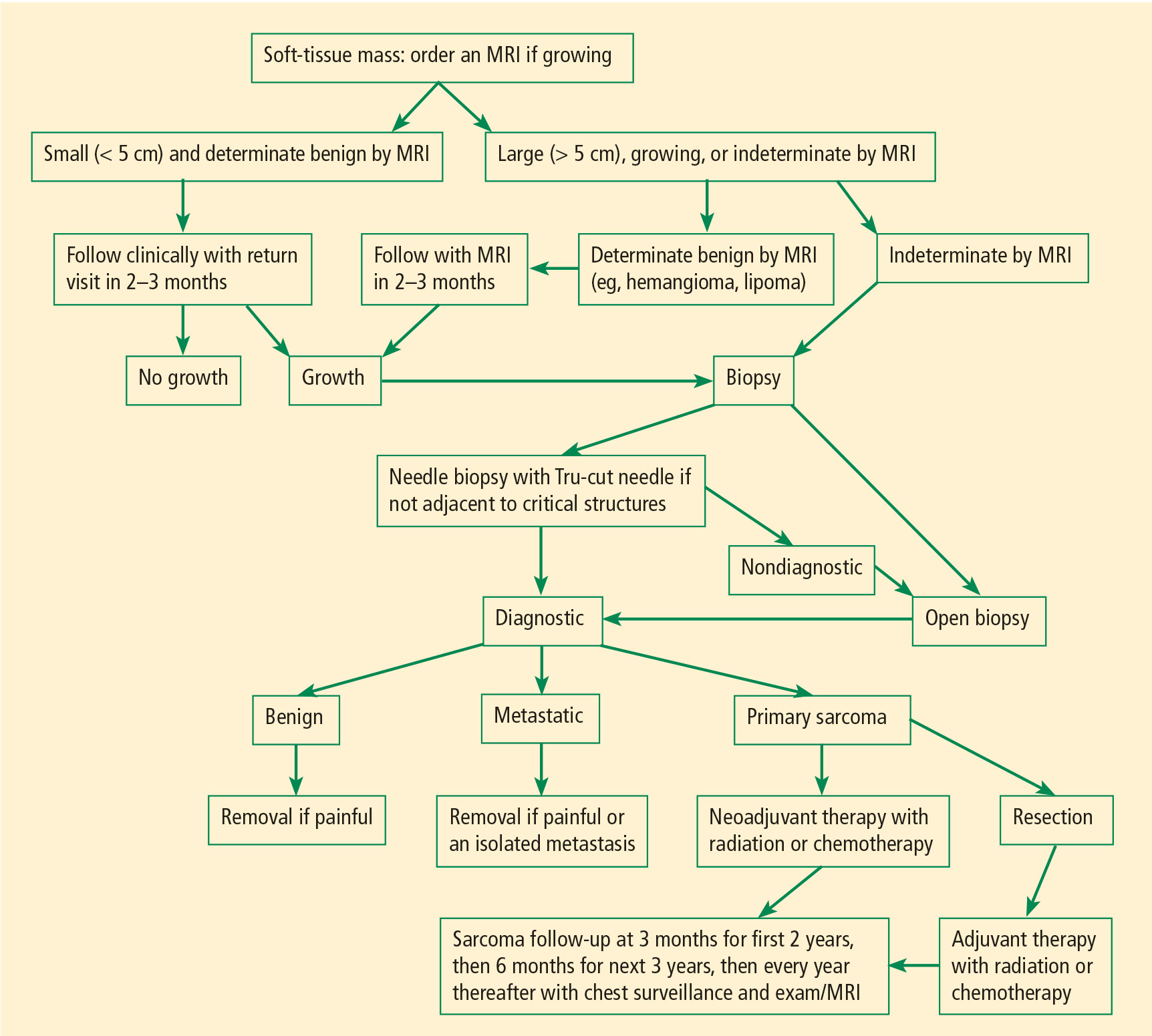
Figure 1 presents in flow chart form our general approach to the evaluation and management of patients with a soft-tissue mass suspicious for sarcoma—an approach detailed in the text below.
History and physical examination
Patients with soft-tissue sarcomas present with a mass that generally is increasing in size. The location and depth of the mass can be assessed on physical examination. In general, the deeper the mass, the more likely it is to be a sarcoma.13 Unlike bone sarcomas, soft-tissue sarcomas frequently are not associated with pain, so lack of pain does not make a mass more likely to be benign. In general, the only way to be sure that a mass is not malignant is to biopsy it. However, there are certain symptoms and signs that make a benign diagnosis much more likely. For example, very soft superficial masses that have not changed in size in years tend to be benign lipomas, and discolored lesions that go away with elevation of the affected body part tend to be hemangiomas.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is the primary imaging method for soft-tissue sarcomas. The benignity of a lesion such as a lipoma or hemangioma may be able to be determined with high certainty on MRI, in which case we call the imaging of the lesion “determinate.” Such lesions with determinate imaging (often referred to as “determinate lesions”) usually do not require a biopsy. However, the nature and identity of most lesions cannot be determined by MRI; although the MRI is still useful to help plan the biopsy in these cases, these lesions are termed “indeterminate” by MRI and should usually be biopsied.
Lesions that can be deemed determinate and usually be diagnosed as benign based on MRI findings include lipomas, hemangiomas, granuloma annulare, and ganglion cysts. However, most other soft-tissue lesions are indeterminate on MRI and, except in rare circumstances, require a biopsy to determine what they are and how they should be treated.