Thrombotic thrombocytopenic purpura: The role of ADAMTS13





ABSTRACTThrombotic thrombocytopenic purpura (TTP) is an uncommon, life-threatening disease requiring prompt diagnosis and initiation of therapeutic plasma exchange to improve patient survival. However, diagnosis is often difficult because of atypical presentations and signs and symptoms that resemble other conditions. Measurements of ADAMTS13 activity, ADAMTS13 inhibitor, and ADAMTS13 autoantibody are useful for diagnosing TTP, guiding therapy, and predicting relapse.
KEY POINTS
- Symptoms of TTP are usually neurologic but can also be cardiac or abdominal. Thrombocytopenia and unexplained microangiopathic hemolytic anemia are sufficient to highly suspect the disease.
- In the appropriate clinical setting, an ADAMTS13 activity level lower than 10% is highly indicative of TTP.
- ADAMTS13 inhibitor and ADAMTS13 antibody assays provide more diagnostic clues. ADAMTS13 antibody is generally absent in the congenital form.
- The ADAMTS13 assay can help distinguish TTP from hemolytic-uremic syndrome, which presents similarly but typically involves normal or only mildly reduced ADAMTS13 activity.
- A strong clinical suspicion of TTP warrants immediate initiation of therapeutic plasma exchange without waiting for ADAMTS13 test results.
A breakthrough in understanding the pathogenesis of thrombotic thrombocytopenic purpura (TTP) came with the discovery of ADAMTS13 (an abbreviation for “a disintegrin and metalloproteinase with thrombospondin type 1 motif, member 13”), a plasma protein that cleaves von Willebrand factor, which interacts with platelets to promote blood clotting. If ADAMTS13 is lacking, unusually large multimers of von Willebrand factor can accumulate and trigger intravascular platelet aggregation and microthrombosis, causing the signs and symptoms of TTP.1–3
This knowledge has practical applications: we can now measure ADAMTS13 activity, ADAMTS13 inhibitor, and antibodies against ADAMTS13 to help us diagnose TTP and distinguish it from other forms of thrombotic microangiopathy, such as hemolytic-uremic syndrome, that have similar symptoms but require different treatment.
Using case studies, this article describes typical presentations of acute and relapsing TTP; the role of laboratory testing, including the ADAMTS13 assay; how to distinguish TTP from other conditions that present similarly; and how to manage this condition.
A HIGH RISK OF DEATH WITHOUT PLASMA EXCHANGE
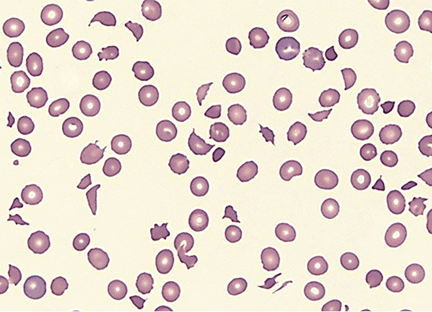
TTP is characterized by disseminated microthrombi composed of agglutinated platelets and von Willebrand factor in small vessels. Tissue damage by microthrombi can cause thrombocytopenia (platelet deficiency), microangiopathic hemolytic anemia (loss of red blood cells caused by destructive conditions in small vessels), and multiorgan failure.1
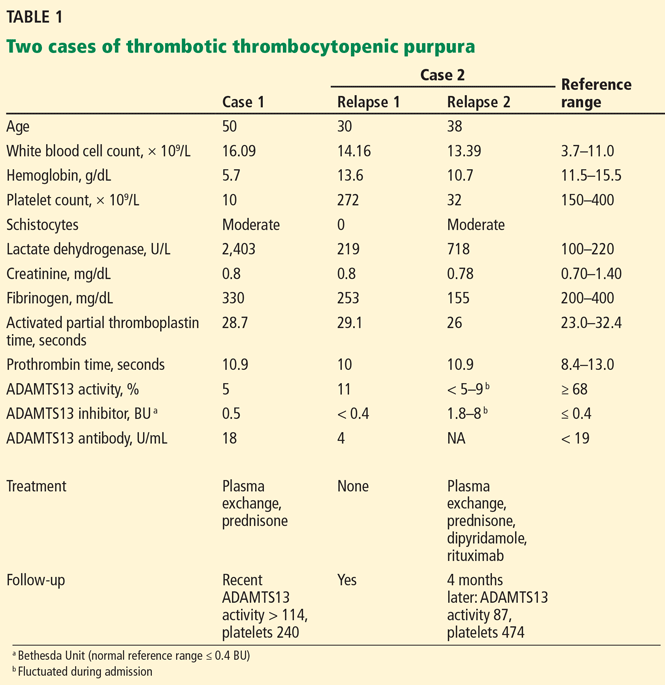
Untreated TTP has a mortality rate of about 90%.1 As shown in Case 1, Case 2, and Table 1, rapid diagnosis and prompt initiation of daily therapeutic plasma exchange can improve this grave outlook.4


ADAMTS13 DEFICIENCY CAN BE ACQUIRED OR CONGENITAL
Two major forms of TTP with ADAMTS13 deficiency and microvascular thrombosis are recognized:
Acquired TTP, the more common form, peaks in incidence between ages 30 and 50.2,5 It more often affects women, particularly during and after pregnancy (its estimated prevalence is 1 in 25,000 pregnancies), and African Americans.6 Acquired TTP may be:
- Primary (idiopathic or autoantibody-mediated), associated with severely decreased ADAMTS13 and the presence of ultra-large von Willebrand factor multimers, or
- Secondary (23%–67% of cases), arising from a variety of conditions, including autoimmune disorders (eg, systemic lupus erythematosus, rheumatoid arthritis), solid organ or hematopoietic cell transplant, malignancy, drugs, and pregnancy (Table 2).1,5–8 Secondary TTP has a worse prognosis than idiopathic TTP.5,9

Congenital TTP (Upshaw-Shulman syndrome) is a rare autosomal-recessive disease caused by compound heterozygous or homozygous mutations of the ADAMTS13 gene, producing nonfunctional ADAMTS13 protein. Patients have severely deficient ADAMTS13 activity but usually do not develop autoantibodies. There is a high risk of chronic, relapsing episodes; identified triggers include pregnancy and heavy alcohol intake.2,10 About half of patients with congenital TTP have an early onset, usually presenting with acute TTP between birth and age 5, and about half have a late onset, usually remaining without symptoms until age 20 to 40.
THE CLINICAL PICTURE OF TTP IS NOT ALWAYS CLASSIC
TTP is primarily diagnosed clinically, but diagnosis is often difficult because of various nonspecific symptoms. Typical TTP presents with the “classic pentad”:
- Severe thrombocytopenia (70%–100% of patients)
- Microangiopathic hemolytic anemia with multiple schistocytes (70%–100%) (Figure 1)
- Neurologic involvement (50%–90%)
- Renal abnormalities (about 50%)
- Fever (25%).
However, the entire picture often does not emerge in a single patient.2,6 Waiting for the entire pentad to develop before diagnosing TTP can have grave clinical consequences,1,2,5 and the presence of thrombocytopenia and unexplained microangiopathic hemolytic anemia are considered clinically sufficient to suspect TTP.5
Neurologic symptoms usually fluctuate. They can include mild abnormalities such as weakness, dizziness, headache, blurred vision, ataxia, and transient mental status changes, as well as severe abnormalities including stroke, seizure, and coma.2,6
Most patients have normal findings on computed tomography and magnetic resonance imaging at the onset of neurologic symptoms or with a history of TTP. Some patients (8%–39%) show reversible acute brain lesions, including ischemic changes.11–13
Other signs and symptoms may result from multiorgan failure due to microthrombosis; ischemia in retinal, coronary, and abdominal circulations; and unconjugated hyperbilirubinemia.2
Atypical presentations. About 18% of patients have cardiac involvement from microvascular occlusion, with arrhythmia, angina, or congestive heart failure. Abdominal pain and pancreatitis occur in 5% to 13%, and visual disturbances in 8% to 10%.
Patients with an atypical presentation may not have laboratory evidence of microangiopathic hemolytic anemia, but an ADAMTS13 assay will show severely decreased activity. Therapeutic plasma exchange can improve atypical symptoms.2,3,10,14,15