Depression and inflammation: Examining the link






Inflammatory conditions may precipitate or perpetuate depression, but the precise relationship is unclear
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
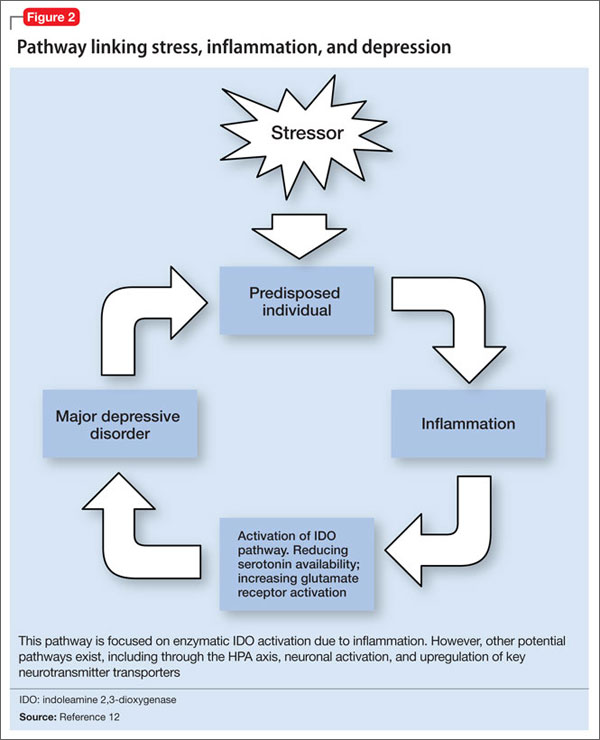
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).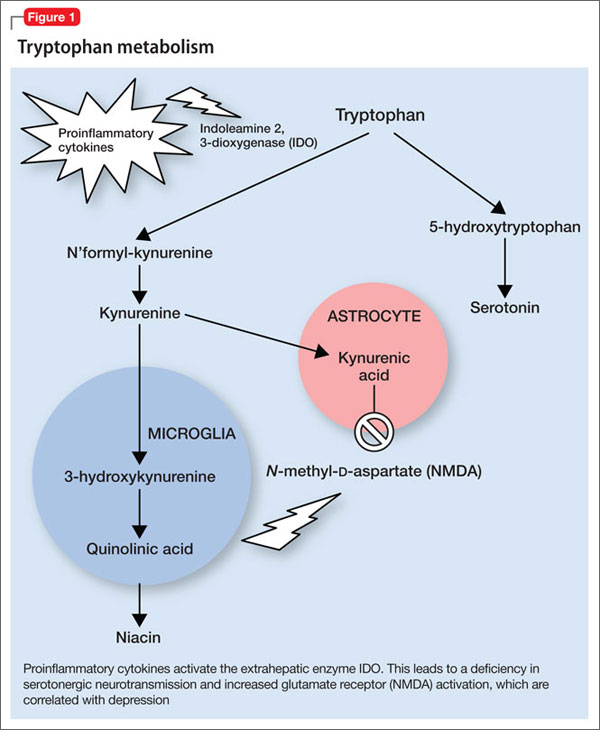
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.