Transvaginal mesh for prolapse: Where are we in 2016?







The ObGyn specialty is moving into a data-driven future for pelvic organ prolapse surgery
In this Article
- Transvaginal mesh and the “Hype Cycle”
- Newer vs older mesh types
- Solutions offered by the Pelvic Floor Disorders Registry
Cheryl B. Iglesia, MD, was the Keynote Speaker at the Pelvic Anatomy and Gynecologic Surgery (PAGS) Symposium held in Las Vegas, Nevada, December 10–12, 2015. This article was developed from Dr. Iglesia's presentation titled "The How, Why and Where of Synthetic and Biologic Mesh: Lessons Learned."
TRANSVAGINAL MESH FOR POP: NEW AND
NOTEWORTHY RESOURCES
• Pelvic Floor Disorders Registry: Overview, registry resources, FAQs, more
• Vaginal mesh manufacturer closing due to lawsuit concerns, Wall Street Journal, 2/29/16
Approximately 300,000 surgeries for pelvic organ prolapse (POP) are performed annually in the United States. In 2006, the peak of synthetic mesh use for prolapse surgery, one-third of all prolapse operations involved some mesh use.1,2 The use of vaginal mesh has declined since the US Food and Drug Administration (FDA) issued warnings in 2008 and 2011.
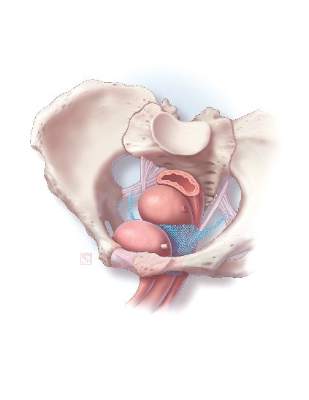
Historically, the use of mesh for gynecologic surgery began in the 1970s, with abdominal POP repair.3 Transvaginal mesh use for POP surgeries became FDA-cleared in 2004. The first cleared mesh device was classified as class II (moderate risk).3 Subsequent mesh devices were given 510(k) clearance, which bypasses clinical trials and requires manufacturers only to show that their product is substantially equivalent to one already on the market.4 More than 40 companies began the manufacturing of mesh devices in the 10 years following the initial cleared device.3
Of course, much controversy has surrounded mesh use in recent years, with common adverse events reported, including severe pelvic pain, pain during intercourse, infection, bleeding, organ perforation, and problems from mesh eroding into surrounding tissues.3 The FDA very recently (in January 2016) reclassified this device from moderate risk to high risk (class III), after indicating in May 2014 that such action was necessary. (See “Timeline of FDA’s actions regarding surgical mesh for pelvic organ prolapse” on page 46.) This reclassification requires a premarket approval application to be filed for each device, with safety and efficacy demonstrated. There are approximately 5 companies currently manufacturing mesh for transvaginal POP repair.3
OBG Management recently sat down with Cheryl Iglesia, MD, director of the Section of Female Pelvic Medicine and Reconstructive Surgery at MedStar Washington Hospital Center and professor in the Departments of Obstetrics/Gynecology and Urology at Georgetown University School of Medicine in Washington, DC. Dr. Iglesia serves, from 2011 through 2017, as a member on the FDA Obstetrics and Gynecology Devices Panel, and she addressed lessons learned over the past decade on synthetic and biologic mesh at the Pelvic Anatomy and Gynecologic Surgery (PAGS) symposium in Las Vegas, Nevada, this past December.
In this Q&A article, she addresses the current state of transvaginal mesh use and how it relates to the innovation adaptation curve (otherwise known as the Hype Cycle), how new mesh types differ from older ones, and how the specialty can move into a future of POP surgery in which innovation and data will rule.
OBG Management: Where is transvaginal mesh use on the so-called “Hype Cycle,” or innovation adaptation curve?
Cheryl B. Iglesia, MD: The Hype Cycle was developed and branded by the Gartner company, an information technology advisory and research firm. This cycle refers to the graphical depictions of how a technology or application will evolve over time. After all, new technologies may make bold promises, and the hype may not translate to commercial viability. Each cycle drills down into the key phases of a technology’s life cycle: the trigger, peak of inflated expectations, trough of disillusionment, slope of enlightenment, and plateau of productivity.5
If we use the Hype Cycle to drill down the phases of transvaginal mesh’s life cycle, we begin in 2004 with the FDA clearance of the first vaginal mesh system (FIGURE).6 The height of its use (the “peak of inflated expectation”) was around 2006, when essentially one-third of all annual surgeries performed for prolapse repair used some type of mesh placed either abdominally or transvaginally.2
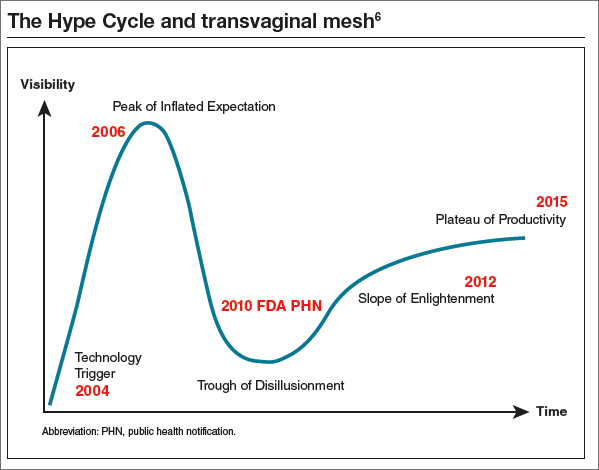
Subsequently, adverse events began being reported to the Manufacturer and User Facility Device Experience (MAUDE) database. In 2008, the FDA published its first notification of serious complications associated with transvaginal placement of surgical mesh, with more than 1,000 reports from 9 surgical mesh manufacturers.7 A second alert followed in 2011.8 By this time, we had reached our “trough of disillusionment.”
In 2016, we have reached the “plateau of productivity” on the innovation adaptation curve. During this phase on the Hype Cycle the criteria for assessing the technology’s viability are clearly defined. I say we are in this phase because now we have a way of completing more postmarket surveillance on mesh devices. We now can see what applying the technology is like in the real world, generalized across many different surgeons’ hands, and we have a way of performing comparative studies with native tissue.