A Strong Diagnosis of Weakness





The approach to clinical conundrums by an expert clinician is revealed through the presentation of an actual patient’s case in an approach typical of a morning report. Similar to patient care, sequential pieces of information are provided to the clinician, who is unfamiliar with the case. The focus is on the thought processes of both the clinical team caring for the patient and the discussant.
© 2017 Society of Hospital Medicine
Because myopathy is most likely, the next step would be to determine if this is an idiopathic inflammatory myopathy, such as polymyositis (PM) or dermatomyositis (DM), secondary inflammatory myopathy, or noninflammatory myopathy due to endocrinopathies. The time course is consistent with an inflammatory myopathy, such as PM or DM. Inclusion body myositis (IBM), another inflammatory myopathy, presents much more insidiously over years and tends to be asymmetric compared to PM. The absence of myalgia, arthralgia, rash, and gastrointestinal symptoms makes myopathy as a component of a connective tissue disease, such as systemic lupus erythematosus, or a mixed connective tissue disease unlikely. The next steps would be laboratory testing of muscle enzymes, complete blood count, biochemical profile, and antinuclear antibody (ANA).
Laboratory studies revealed a white blood cell count of 4460/mm3 with normal differential, hemoglobin 12.5 g/dL, and platelet count 345,000/mm3. Creatinine was 0.87 mg/dL, aspartate aminotransferase 61 IU/mL, alanine aminotransferase 45 IU/mL, and creatine kinase (CK) 529 U/L (normal range, 38-174 U/L). Other liver function enzymes were normal. Biochemistry studies disclosed normal sodium, potassium, glucose, calcium, and magnesium levels. Dipstick urinalysis revealed blood and protein, and the microscopic examination of urinary sediment was unremarkable without the presence of erythrocytes. Twenty-four-hour creatinine clearance was 106 mL/min (normal range, 97-137 mL/min). Chest radiography was unrevealing.
The modest increase in CK, evidence of myoglobinuria, and proteinuria can all occur with an inflammatory or metabolic myopathy. The combination of proximal and distal weakness, coupled with only a modestly elevated CK, makes IBM more likely than PM, as PM usually presents with proximal weakness and much higher CK values. Normal skin examination makes DM less likely, as skin manifestations are generally found at time of presentation. The onset of symptoms after age 50 and the patient being male also favor IBM, though a longer time course would be expected. Definitively distinguishing IBM from PM is important because treatment and prognosis differ.
Thyroid function and HIV testing should be obtained. ANA, more common in PM than in IBM, should be checked because these myopathies can be associated with other autoimmune diseases. Imaging is generally not essential, although magnetic resonance imaging (MRI) of the thighs may help to differentiate IBM from PM. Electromyography (EMG) should be done to determine the pattern of myopathy and select muscle biopsy sites.
Additional testing revealed a normal thyroid stimulating hormone level. HIV and ANA were negative. Serum aldolase level was 19 IU/L (normal range, 2.7-5.9 IU/L), myoglobin 277 ng/mL (normal range, 28-72 ng/mL), lactate dehydrogenase 416 IU/mL (normal range, 119-229 IU/mL), and C-reactive protein 0.32 mg/dL. An EMG revealed mild myogenic changes in all extremities. An MRI of the left brachial muscle revealed multiple scattered high-signal lesions.
The EMG and MRI findings are consistent with an inflammatory myopathy. The modest elevation in muscle enzymes and negative ANA are more consistent with IBM since most patients with PM or DM are ANA positive. Muscle biopsy can be very helpful in establishing the etiology of myopathy.
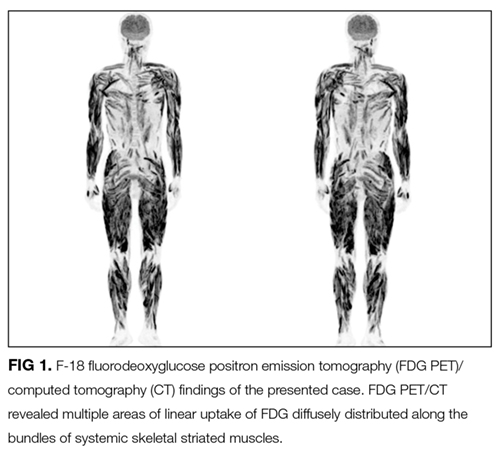
Malignancy is associated with DM and PM in about 9% and 4% of patients, respectively. The common cancers associated with these conditions are adenocarcinomas of the ovary, cervix, lung, pancreas, and stomach. Most cancers are diagnosed around the time of myositis diagnosis, although they can precede or follow by years. Idiopathic IBM is not associated with cancer.
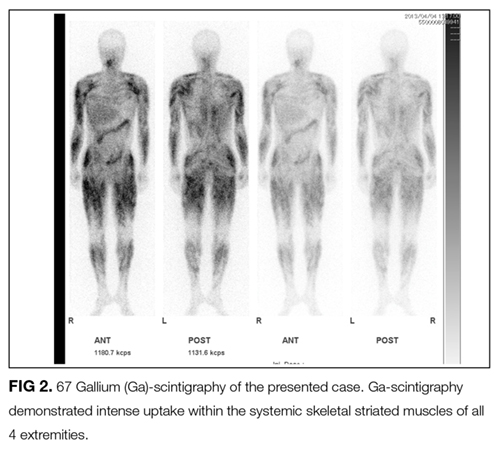
Open surgical muscle biopsy of the left biceps brachii was performed. Light microscopic examination disclosed interstitial edema and noncaseating granulomas. Immunostaining revealed an increase in the number of cluster of differentiation (CD) 4+ T cells. Caseating granulomas and Langhans giant cells were not present (Figure 3).

Tuberculin reaction and interferon-γ-release assay were negative. Staining for AFB and fungi was negative. ANCA, rheumatoid factor (RF), anti-Ro/SSA, anti-La/SSB, anti-Sm, anti-RNP, and anti-Jo-1 were all negative or unremarkable. Serum angiotensin converting enzyme (ACE) level was 155.6 U/L (normal range, 7-25 U/L). Twenty-four-hour urine analysis revealed calcium excretion of 517.7 mg/day (normal range, 58-450 mg/day), β2-microglobulin 69,627 ug/day (normal range, <254 ug/day), and N-acetyl-D-glucosamine 95.3 U/day (normal range, <5.1 U/day) with a normal creatinine clearance. Serum intact parathyroid hormone level (PTH) was 5 pg/mL (normal range, 10-65 pg/mL), and 25-hydroxyvitamin D level was 51.1 ng/mL (normal range, 30-80 ng/mL). A CT of the thorax revealed a small ground-glass density lesion in the left lower lobe but no hilar or mediastinal lymphadenopathy.
Negative ANCA, RF, and autoantibodies exclude systemic vasculitis and connective tissue disease as causes of GM. Hypercalciuria is suggestive of granulomatous production of calcitriol, which, in turn, suppresses PTH. Hypercalcemia is not common in patients with sarcoidosis, but hypercalciuria occurs frequently. Serum ACE is a marker associated with sarcoidosis, but its diagnostic and prognostic utility is unclear.