Von Willebrand Disease: Approach to Diagnosis and Management





Introduction
von Willebrand disease (VWD) is an inherited bleeding disorder caused by deficient or defective plasma von Willebrand factor (VWF). VWF is an adhesive multimeric plasma glycoprotein that performs 2 major functions in hemostasis: it mediates platelet adhesion to injured subendothelium via glycoprotein 1bα (GPIbα), and it binds and stabilizes factor VIII (FVIII) in circulation, protecting it from proteolytic degradation by enzymes. The current VWD classification recognizes 3 types (Table 1).1 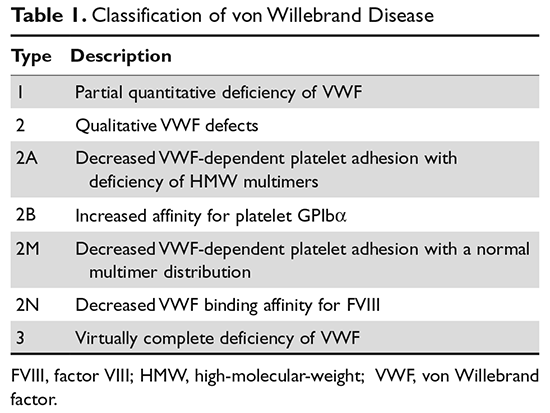
Prevalence
VWD is the most common inherited bleeding disorder. However, because VWF levels are highly variable and disease severity ranges from mild bleeding symptoms to severe or life-threatening bleeds, the reported prevalence of VWD depends on the diagnostic definition used. Two large epidemiologic studies have reported prevalence rates of approximately 1%.2,3 In these studies, healthy school-aged children were screened and diagnosed with VWD based on low VWF activity, measured as ristocetin cofactor, and a personal and family history of bleeding symptoms. At the other extreme, when considering patients whose bleeding symptoms are sufficiently severe to warrant referral to specialized centers, the reported prevalence of VWD ranges from 20 to 113 per million.4 These studies likely over- and underestimate clinically significant VWD. More recent studies suggest that the prevalence of VWD in individuals whose bleeding symptoms are significant enough to present to a primary care physician is approximately 0.1%.5 This figure is likely a more accurate estimate of the true prevalence of symptomatic VWD.
Although VWD is autosomally inherited, females are more likely to present with bleeding symptoms and be diagnosed because of increased exposure to bleeding challenges, such as menorrhagia and childbirth. VWD does not show any geographic or ethnic predilection, but there is an increased prevalence of the recessive forms, such as type 2N and type 3 VWD, in areas with high rates of consanguinity.
VWF Protein Structure and Function
The VWF gene is located on chromosome 12 at p13.3 and spans 178 kb comprising 52 exons.6 The expression of the VWF gene is tightly restricted to endothelial cells, platelets, and megakaryocytes, where VWF is stored in Weibel-Palade bodies and α-granules. VWF is a large multimeric glycoprotein with several important functional domains (Figure). 
Classification, Pathophysiology, and Genetics
The International Society of Thrombosis and Hemostasis (ISTH) classification of VWD was updated in 2006 (Table 1).1 It incorporates important aspects of clinical phenotype, pathophysiological mechanisms, and treatment considerations. The 3 categories are: type 1, which is a partial quantitative deficiency; type 2 with 4 subtypes (2A, 2B, 2M, and 2N), which is a qualitative defect; and type 3, which is a virtual absence of VWF. Although the diagnosis and categorization of VWD can be achieved with widely available laboratory testing, further subcategorization among type 2 VWD subtypes may require referral to a specialized laboratory. The current ISTH classification intentionally does not incorporate genotypic data. In type 2 or type 3 VWD disease, VWF mutations are identified in more than 90% of cases and are completely penetrant, whereas mutations are identified in only approximately 65% of type 1 VWD cases and have been associated with incomplete penetrance and variable expressivity.10 These studies suggest that type 1 VWD is an oligogenic disease with mutations in genes regulating secretion or clearance contributing to a VWD phenotype.