Current Therapeutic Approaches to Renal Cell Carcinoma





INTRODUCTION
Renal cell carcinoma (RCC) is the most common malignancy arising in the kidney, comprising 90% of all renal tumors.1 Approximately 55,000 new RCC cases are diagnosed each year.2 Patients with RCC are often asymptomatic, and most cases are discovered as incidental findings on abdominal imaging performed during evaluation of nonrenal complaints. Limited-stage RCC that is found early can be cured surgically, with estimated 5-year survival rates approaching 90%; however, long-term survival for metastatic disease is poor, with rates ranging from 0% to 20%.2 Advanced RCC is resistant to conventional chemotherapy and radiotherapy, and outcomes for patients with metastatic or unresectable RCC remain poor. However, the recent development of new therapeutic modalities that target tumor molecular pathways has expanded the treatment options for these patients and changed the management of RCC.
EPIDEMIOLOGY AND CLASSIFICATION
Median age at diagnosis in the United States is 64 years. Men have a higher incidence of RCC than women, with the highest incidence seen in American Indian and Alaska Native men (30.1 per 100,000 population). Genetic syndromes account for 2% to 4% of all RCCs.2 Risk factors for RCC include smoking, hypertension, obesity, and acquired cystic kidney disease that is associated with end-stage renal failure.3 Longer duration of tobacco use is associated with a more aggressive course.
The 2004 World Health Organization (WHO) classification of renal tumors summarizes the previous classification systems (including the Heidelberg and Mainz classification systems) to describe different categories of RCC based on histologic and molecular genetics characteristics.2 Using the WHO classification criteria, RCC comprises 90% of all renal tumors, with clear cell being the most common type (80%).2 Other types of renal tumors include papillary, chromophobe, oncocytoma, and collecting-duct or Bellini duct tumors. Approximately 3% to 5% of tumors are unclassified. Oncocytomas are generally considered benign, and chromophobe tumors typically have an indolent course and rarely metastasize. Sarcomatoid differentiation can be seen in any histologic type and is associated with a worse prognosis. While different types of tumors may be seen in the kidney (such as transitional cell or lymphomas), the focus of this review is the primary malignancies of the renal parenchyma.
,FAMILIAL SYNDROMES
Several genetic syndromes have been identified by studying families with inherited RCC. Among these, von Hippel-Lindau (VHL) gene mutation is the most commonly found inherited genetic defect. Table 1 summarizes the incidence of gene mutations and the corresponding histologic appearance of the most common sporadic and hereditary RCCs.4
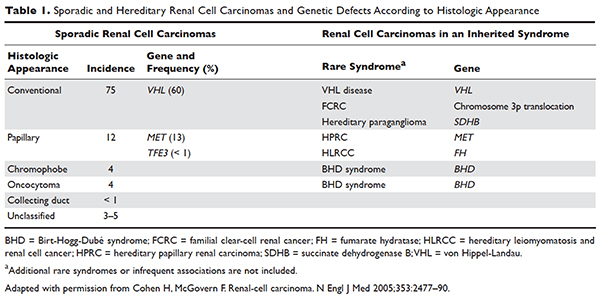
VHL disease is an autosomal dominant familial syndrome. Patients with this mutation are at higher risk for developing RCC (clear cell histology), retinal angiomas, pheochromocytomas, as well as hemangioblastomas of the central nervous system (CNS).4 Of all the genetic mutations seen in RCC, the somatic mutation in the VHL tumor-suppressor gene is by far the most common.5 VHL targets hypoxia–inducible factor-1 alpha (HIF-α) for ubiquitination and subsequent degradation, which has been shown to suppress the growth of clear-cell RCC in mouse models.6–8 HIF expression under hypoxic conditions leads to activation of a number of genes important in blood vessel development, cell proliferation, and glucose metabolism, including vascular endothelial growth factor (VEGF), erythropoietin, platelet-derived growth factor beta (PDGF-β), transforming growth factor alpha (TGF-α), and glucose transporter-1 (GLUT-1). Mutation in the VHL gene prevents degradation of the HIF-α protein, thereby leading to increased expression of these downstream proteins, including MET and Axl. The upregulation of these angiogenic factors is thought to be the underlying process for increased vascularity of CNS hemangioblastomas and clear-cell renal tumors in VHL disease.4–8
Other less common genetic syndromes seen in hereditary RCC include hereditary papillary RCC, hereditary leiomyomatosis, and Birt-Hogg-Dubé (BHD) syndrome.9 In hereditary papillary RCC, the MET gene is mutated. BHD syndrome is a rare, autosomal dominant syndrome characterized by hair follicle hamartomas of the face and neck. About 15% of patients have multiple renal tumors, the majority of which are of the chromophobe or mixed chromophobe-oncocytoma histology. The BHD gene encodes the protein folliculin, which is thought to be a tumor-suppressor gene.
DIAGNOSIS AND STAGING
CASE PRESENTATION
A 74-year-old man who works as an airplane mechanic repairman presents to the emergency department with sudden worsening of chronic right upper arm and shoulder pain after lifting a jug of orange juice. He does not have a significant past medical history and initially thought that his pain was due to a work-related injury. Upon initial evaluation in the emergency department he is found to have a fracture of his right humerus. Given that the fracture appears to be pathologic, further work-up is recommended.