Hemophilia A and B: An Overview





INTRODUCTION
Hemophilia A and B are the most common severe inherited bleeding disorders. The incidence of hemophilia is 1 in 5000 live male births, with hemophilia A occurring 4 times more commonly than hemophilia B. The associated decrease in factor VIII in hemophilia A was initially identified in 1947, and the decrease in factor IX associated with hemophilia B was identified 5 years later.1,2 Both conditions are inherited as X-linked recessive traits. Queen Victoria of Britain, who reigned from 1837 to 1901, was a carrier of hemophilia and had 2 carrier daughters, Alice and Beatrice, and a son with hemophilia, Leopold.3 In 1984 and 1985, the genes for factor VIII and factor IX were cloned, and in 1989 recombinant factor VIII was first used clinically.4–7
PATHOPHYSIOLOGY
Both factors VIII and IX are crucial for normal thrombin generation in the coagulation pathway. After any injury, the initial hemostatic event is the formation of a platelet plug. Once the platelet plug is formed, subsequent generation of fibrin prevents continued oozing from the affected site. In hemophilia A and B, the propagation phase of coagulation is impaired, and as a result, the formation of clot is delayed and is not robust. Due to the delayed formation of an abnormal clot, patients with hemophilia do not bleed rapidly but rather ooze continuously. Rebleeding is a common occurrence in inadequately treated patients.8
GENETICS
The gene for factor VIII (F8) is located in the most distal band (Xq28) of the long arm of the X chromosome. Spanning more than 186 kb, it is one of the largest genes known.9,10 The gene for factor IX (F9) is located at Xq27.1 and spans 33 kb.7 Defects in the F8 gene associated with hemophilia A may be divided into several categories: gross gene rearrangements; insertions or deletions of genetic sequence of a size varying from 1 base pair up to the entire gene; or single DNA base substitutions resulting in either amino acid replacement (missense), premature peptide chain termination (nonsense, or stop mutations), or mRNA splicing defects. All classes of defects can result in severe disease. However, the single most clinically important defect is a gene rearrangement (an inversion) involving F8 intron 22, which results in approximately 50% of all severe hemophilia A cases worldwide.11,12 In hemophilia B, point mutations are by far the most common type of abnormality. Generally, they are caused by DNA polymerases adding the wrong nucleotide during replication.13
,HEMOPHILIA IN FEMALES
X-Inactivation (also called Lyonization) is a process that occurs early in embryonic development in female mammals where 1 of the 2 copies of the X chromosome present is inactivated; it is the reason why some female carriers of hemophilia can become symptomatic. Approximately one third of carriers have clotting factor levels of less than 60% of normal and may experience abnormal bleeding.14,15 In most cases, carriers experience symptoms similar to those seen in men with mild hemophilia, as well as some that are specific to women. Symptomatic carriers and women with hemophilia may bruise more easily; may experience prolonged bleeding after surgery; may experience serious bleeding after trauma; often have heavier and more prolonged bleeding during their periods (menorrhagia) and are more likely to require an iron supplement or to undergo hysterectomy; and are more likely to have postpartum bleeding following childbirth.14,15
CLINICAL MANIFESTATIONS
Hemorrhage in patients with hemophilia may occur with minimal or unknown trauma. Patients with severe hemophilia (factor level of < 1 IU/dL or < 1% of normal) often experience spontaneous bleeding into joints or muscles. Those with moderate hemophilia (factor level of 1–5 IU/dL or 1%–5% of normal) seldom experience spontaneous hemorrhage and usually have prolonged bleeding with minor trauma or surgery. Patients with mild hemophilia (factor level > 5 IU/dL but less than 40 IU/dL or > 5% but < 40% of normal) experience severe hemorrhage only following moderate to severe trauma or surgery, and rarely experience spontaneous bleeding. Depending on the site, bleeding can be serious (joints; muscles, especially deep compartments [iliopsoas, calf, and forearm]; mucous membranes in the mouth, gums, nose, and genitourinary tract) or life-threatening (intracranial, neck/throat, gastrointestinal). The joints and muscles are the most common sites of bleeding (Table 1).

MUSCULOSKELETAL BLEEDING
The hallmark of hemophilia is deep bleeding into the joints and muscles. Without prophylactic factor treatment, patients with severe hemophilia A or B may have a bleeding episode as often as once or twice a week. Hemarthrosis episodes typically begin when the child reaches the toddler age. One of the first signs of hemarthrosis is a tingling sensation and feeling of warmth which is soon followed by pain and decreased range of motion of the joint as a result of distension of the joint capsule. Prompt, aggressive treatment with factor replacement therapy is the key to prevent further bleeding and minimize potential long-term complications. Severe chronic arthropathy may develop in older children and adults who have not received aggressive treatment (Figure).
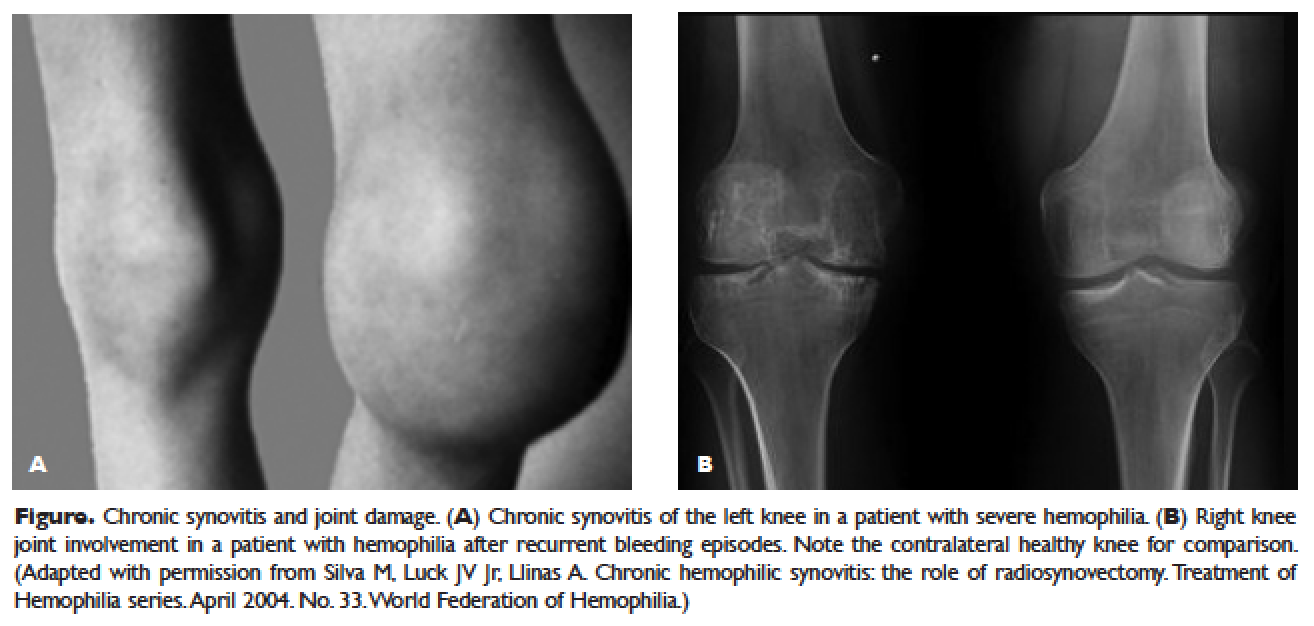
Bleeding into the muscle can manifest as a vague feeling of pain on motion. Swelling may not be obvious and the mass may be difficult to palpate, although the circumference of the affected limb will be increased. Among the muscle bleeds, iliopsoas bleed deserves a special mention because of its potential to cause life-threatening hypovolemic shock as large volumes of blood can be lost into the retroperitoneal space. These patients present with vague abdominal pain or upper thigh discomfort. The hip is flexed and outwardly rotated. The diagnosis is confirmed by computed tomography (CT) or ultrasound.