Intravascular large B-cell lymphoma: an elusive diagnosis with challenging management





Accepted for publication November 2, 2018
Correspondence Akash Mukherjee, MD; AMukherjee21@mdanderson.org
Disclosures The authors report no disclosures/conflicts of interest.
©2018 Frontline Medical Communications
doi https://doi.org/10.12788/jcso.0425
Intravascular large B-cell lymphoma (IVBCL) is an aggressive and systemically disseminated disease that affects the elderly, with a median age of diagnosis around 70 years and no gender predilection. It is a rare subtype of extranodal diffuse large B-cell lymphoma (DLBCL) characterized by selective growth of neoplastic cells within blood vessel lumen without any obvious extravascular tumor mass. Hence, an absence of marked lymphadenopathy and heterogeneous clinical presentation make it difficult to diagnose accurately and timely, with roughly half of the cases found postmortem in previous case reports.1,2 The exact incidence of this disease is not known, but more recently, the accuracy of diagnosis of this type of lymphoma has improved with random skin and bone marrow biopsy.1,2 We present here a clinical case of this disease with an atypical presentation followed by a detailed review of its clinical aspects.
Case presentation and summary
A 43-year-old white woman with a history of hypothyroidism and recurrent ovarian cysts presented to clinic with 3 months of loss of appetite, abdominal distension, pelvic pain, and progressive lower-extremity swelling. A physical examination was notable for marked abdominal distension, diffuse lower abdominal tenderness, and pitting lower-extremity edema. No skin rash or any other cutaneous abnormality was noted on exam. Laboratory test results revealed a lactate dehydrogenase (LDH) level of 1652 U/L and a CA-125 level of 50 U/mL (reference range, 0-35 U/mL). No significant beta-human chorionic gonadotropin and alpha-fetoprotein levels were detected. Computed-tomographic (CT) imaging revealed small bilateral pleural effusions and gallbladder wall thickening with abdominal wall edema, but it was otherwise unrevealing. An echocardiogram showed normal cardiac structure and function, with a left ventricular ejection fraction of 60%. No protein was detected in the patient’s urine, and thyroid function tests were unrevealing. Doppler ultrasound studies of her lower extremities and abdomen revealed no thrombosis. Given the patient’s continued pelvic pain, history of ovarian cysts, and elevation in CA-125, she underwent a laparoscopic total abdominal hysterectomy and bilateral salpingoopherectomy.
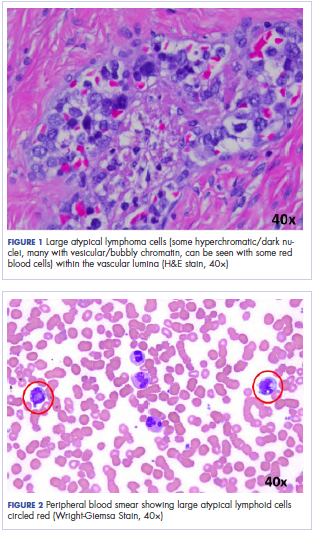
Histologic examination revealed neoplastic cells involving only the vascular lumina of the cervix, endomyometrium, bilateral fallopian tubes, and bilateral ovaries (Figure 1). Immunohistochemistry stains were positive for CD5, CD20, PAX-5, CD45, BCL-2, and BCL-6 and focally positive for CD10. Peripheral smear showed pseudo-Pelger–Huet cells with 5% atypical lymphoma cells (Figure 2). Complete staging with positron-emission and CT (PET–CT) imaging revealed no metabolic activity, and a bone marrow biopsy showed trilineage hematopoiesis with adequate maturation and less than 5% of the marrow involved with large B-cell lymphoma cells. A diagnosis of IVBCL was made.
,
Further work-up to rule out involvement of the central nervous system (CNS) included magnetic-resonance imaging (MRI) of the brain and cerebrospinal fluid (CSF) cytology and flow cytometry, which were negative.
Our patient underwent treatment with 6 cycles of infusional, dose-adjusted R-EPOCH (rituximab, etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) and 6 doses of prophylactic intrathecal chemotherapy with alternating methotrexate and cytarabine (Ara-C), and initial and subsequent CSF sampling showed no disease involvement. Consolidation with high-dose chemotherapy with R-BEAM (rituximab, carmustine, etoposide, Ara-C [cytarabine], melphalan) followed by rescue autologous stem cell transplantation (ASCT) was performed, and the patient has remained in clinical and hematologic remission for the past 24 months.