Expanding treatment options for diverse neuroendocrine tumors





Citation JCSO 2017;15(6):e339-e345
©2017 Frontline Medical Communications
doi https://doi.org/10.12788/jcso.0383
Submit a paper here
Neuroendocrine tumors (NETs) are an extremely diverse group of cancers that have steadily increased in incidence in recent years. They can prove challenging to treat but, as we discuss here, a steady evolution in our understanding of NETs has significantly expanded the scope of therapeutic options.
A unique tumor type
NETs arise from neuroendocrine cells – cells with features of both nerve and endocrine cells that have important physiological functions, including the production and release of hormones. These tumors were first recognized by a German pathologist in the mid-1800s and were initially referred to as carcinoids in reference to their carcinoma-like appearance but lack of other malignant features.1
Unlike other solid tumors, which are associated with a particular primary location, NETs can arise anywhere in the body where neuroendocrine cells are found. They are also unique in their ability to oversecrete bioactive substances that regulate bodily functions, which results in an associated clinical syndrome, known as carcinoid syndrome, in up to 35% of patients.2,3
Although they are considered to be a relatively rare type of tumor, the incidence of NETs has been increasing in recent years. According to data from the Surveillance, Epidemiology and End Results (SEER) program, the age-adjusted incidence of NETs increased more than two-and-a-half fold during 1973-2004 and the rise is predicted to continue at an accelerated rate.4
Historically, NETs have been thought of as relatively benign because of their slow-growing nature, but it is now widely appreciated that they often metastasize. Furthermore, many patients are not symptomatic at first, so around half of all cases are not diagnosed until they have reached this more aggressive stage.4
The challenge of NET diversity
The most common type of NETs are those that arise in the gastrointestinal tract (GI-NET), representing more than 65% of cases, and for which the “carcinoid” terminology often is still applied. GI-NETs most frequently arise in the small intestine (41.8%), rectum (27.4%), and stomach (8.7%).4,5
About a quarter of NETs originate in the bronchopulmonary system, including the lungs and the thymus. Thymic NETs are particularly aggressive and are associated with a poor prognosis. Pancreatic NETs (pNETs) make up the next largest group, although they represent less than 1% of total NETs. Compared with the most common type of pancreatic cancer, pancreatic ductal adenocarcinoma, they have a more favorable prognosis. pNETs are often grouped together with GI-NETs and referred to as gastroenteropancreatic NETs (GEP-NETs).3-5 Other rarer types of NET include Merkel cell carcinoma (a type of skin cancer) and medullary thyroid cancers.
The classification network
NETs are classified according to the anatomic site from which they originate, as well as their histology, grade, and stage. Another important consideration is their level of hormone secretion. “Functional” and “nonfunctional” NETs both produce hormones, but only the former cause related symptoms.3,4,6
Functionality plays a particularly important role in the subclassification of GEP-NETs. Functional pNETs, for instance, are further divided according to the clinical syndromes associated with the hormones they produce, as insulinomas, glucagonomas, gastrinomas, somatostatinomas, and VIPomas (producing vasoactive intestinal peptide).7,8
In 2010, the World Health Organization developed a classification system for GEP-NETs that categorized these tumors as well differentiated (grade 1 or 2, depending on their rate of proliferation) and poorly differentiated (grade 3).9 The WHO classification of bronchopulmonary NETs, published in 2015, is slightly different; broken down into 3 subgroups, typical carcinoid, atypical carcinoid (corresponding to grade 1 and 2 GEP-NETs), and large and small-cell NETs (equivalent to grade 3 GEP-NETs).10
Although NETs develop from the same cell type, they in fact comprise a spectrum of diseases that vary extensively in their underlying biology, histology, and clinical behavior. Both the diversity and unique nature of NETs have become increasingly evident in recent years with the application of next-generation sequencing technologies to this tumor type. In general, NETs seem to be more genetically stable than other tumor types from the same primary location, and have fewer somatic mutations. The classic tumor suppressors and oncogenes that drive other tumor types are not common in NETs.6,11
The diversity of NETs presents a diagnostic and therapeutic challenge and, until recently, there was a paucity of effective treatment options. In the past decade, an evolution in our understanding of the molecular mechanisms underlying these tumors has altered the treatment landscape for well-differentiated tumors as an expanding array of targeted therapies with proven efficacy have become available (Table 1).
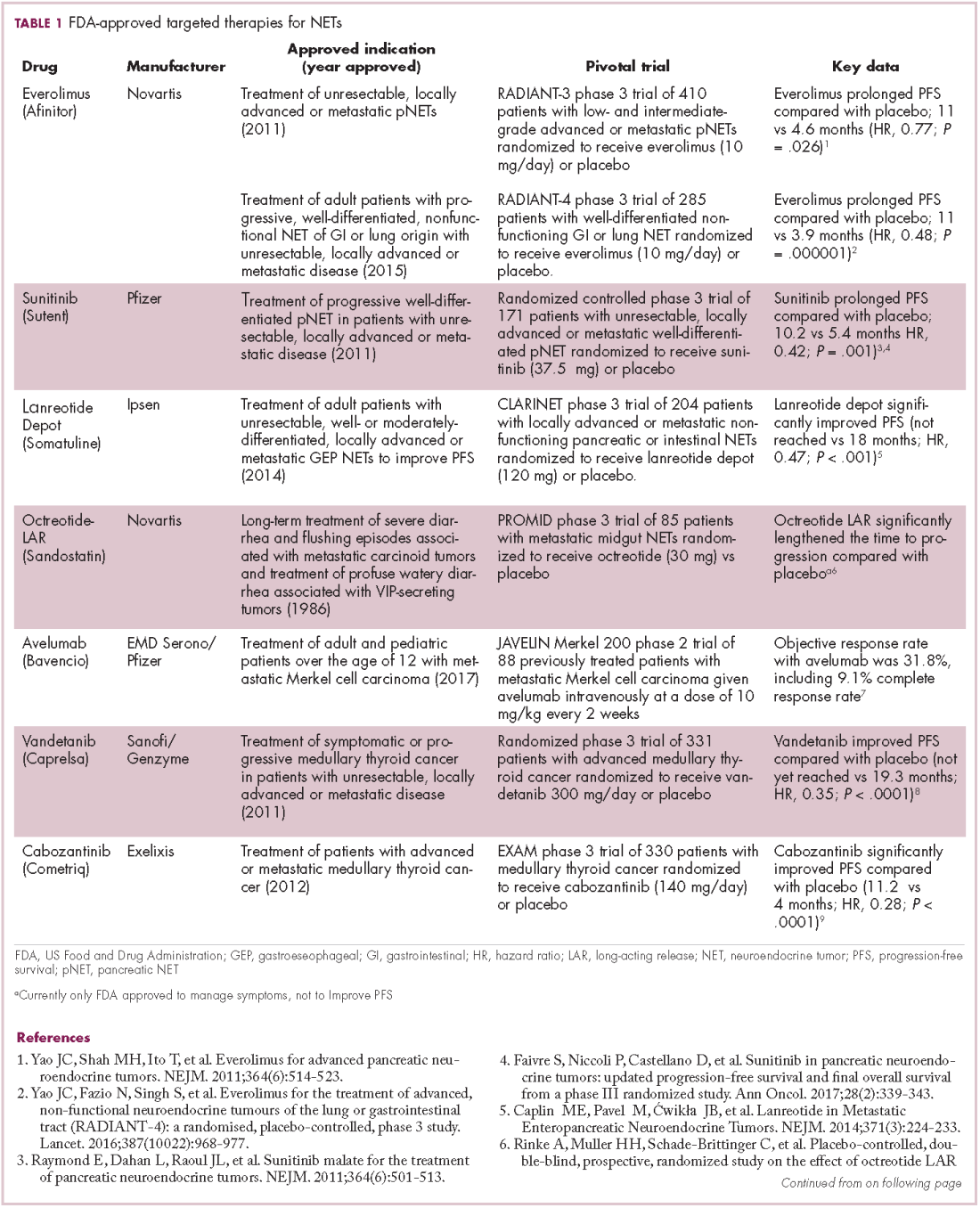

Their poorly differentiated counterparts, on the other hand, continue to present a significant unmet need.
Somatostatin analogs lead the charge
The fact that many NETs overexpress hormone receptors presents a significant therapeutic opportunity, and among the most successful targets to date are the somatostatin receptors (SSTRs). There are 5 main SSTRs that each bind to somatostatin with different effects on cell signaling and expression that varies according to the type of NET.