Cardiac failure due to left atrial angiosarcoma






Abstract
Primary heart sarcomas are rare and represent 20% of all primary cardiac tumors. Symptoms depend on which chambers and cardiac structures are involved. Angiosarcoma is one of the most common and the most aggressive types of primary heart sarcomas. Typically, these tumors are found in the right atrium, however, cardiac angiosarcomas may involve any part of the heart. Most of these tumors are diagnosed in advanced stages and the patient prognosis is poor. Most tumors are diagnosed using echocardiography. Computed tomography (CT) and magnetic resonance imaging (MRI) provide useful information on tumor size and location for planning surgery, which is the only treatment shown to increase survival. We present the case of a 69-year-old woman who presented to the emergency department with hypotension, dyspnea and progressive shortness of breath. After adequate resuscitation, a cardiac mass was identified and surgery was successfully performed. Pathology confirmed a grade 2 primary heart angiosarcoma. Following surgery, the patient was admitted to the intensive care unit and later died secondary to multi-organ system failure.
Introduction
Primary heart angiosarcoma is an aggressive and usually fatal cardiac neoplasm (1). Angiosarcomas can originate at any location in the heart (2, 3), but these tumors typically reside in the right atrium and frequently cause nonspecific symptoms such as dyspnea, cough, heart failure, and arrhythmias. (2) Surgery followed by chemotherapy is the typical approach to these tumors. (4)
We present the case of a 69-year-old woman who presented to the emergency department with hypotension and severe dyspnea.
Case Report
The patient was a 69-year-old woman with a medical history of diabetes. A week before seeking care in the emergency department, she experienced a general feeling of unwellness, dyspnea, and mild respiratory distress. She reported these symptoms had become more and more severe in the last 24 hours and were accompanied by acute chest pain and progressive shortness of breath.
On clinical examination, the patient was hypotensive, had tachypnea and tachycardia, and was hypoxic. Cardiac auscultation detected a systolic murmur in the apex, and auscultation of the lungs revealed crackles and rales, especially at the bases of the lungs. The remainder of her clinical examination was unremarkable. She had sinus tachycardia on an electrocardiogram. A chest X-ray showed a left atrial enlargement along with some patchy opacities in the middle and lower zones of the lungs, along with Kerley B lines suggestive of pulmonary edema.
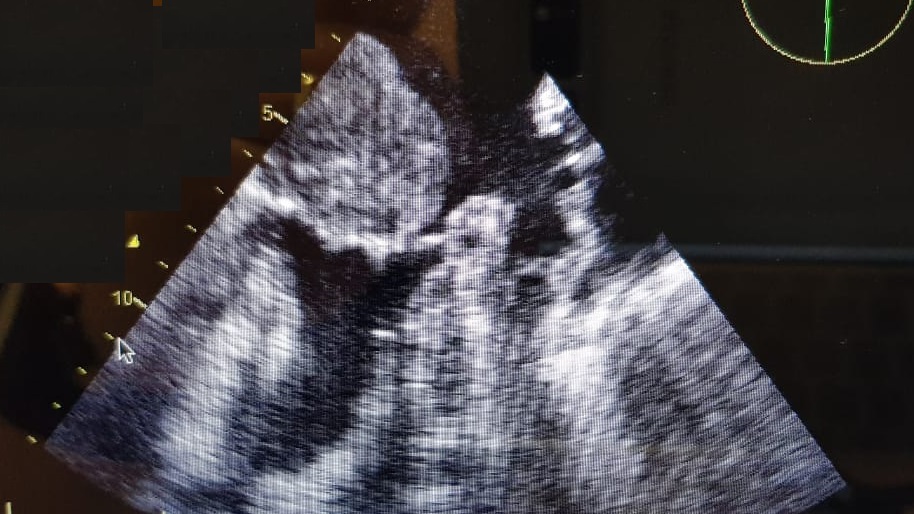
With these findings, and after adequate resuscitation, a contrast-enhanced computed tomography (CT) scan detected a filling defect in the left atrium suggestive of a large intra-cardiac mass with a thick and hyper-enhanced interatrial septum. Bilateral pleural effusions also were evident, (Figure 1A) hence an echocardiogram was requested and it confirmed the presence of a 30 x 29 x 40 mm lobulated highly mobile mass in the left atrium.
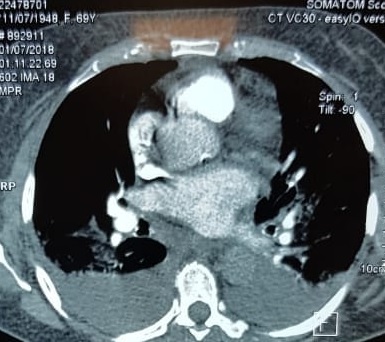
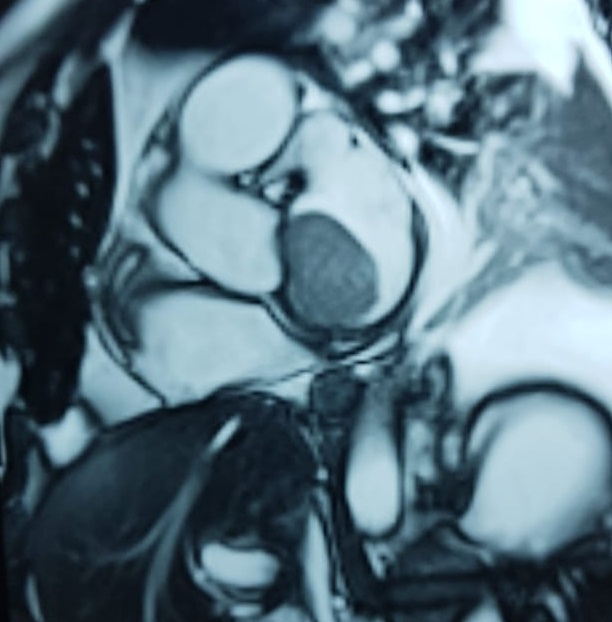
After a cardiothoracic consultation, cardiac magnetic resonance imaging (MRI) was performed. The findings showed the presence of a 58 x 45 x 6 mm well-circumscribed hyperemic mass on the anterior leaflet of the mitral valve and a second 10 x 10 x 6 mm smaller mass firmly adhered to the posterior leaflet of the mitral valve.
The patient, who was hypotensive and hypoxic, was admitted to the hospital for surgical treatment.
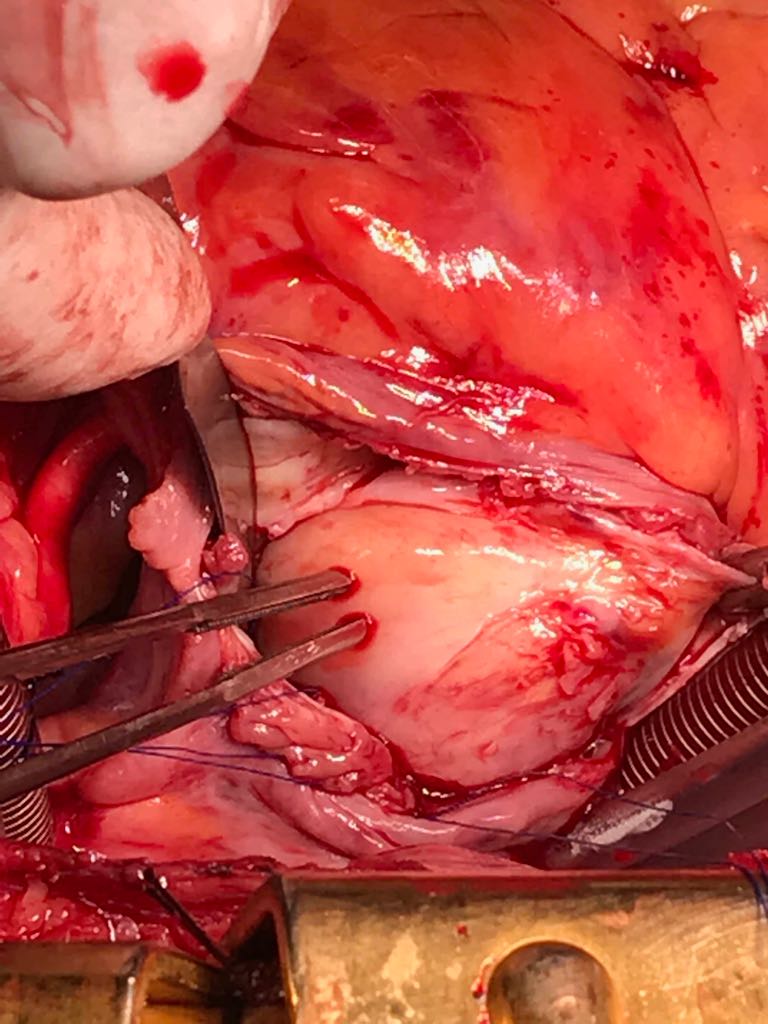
Following sternotomy and cardiopulmonary bypass, a right atriotomy was performed using a trans-septal approach. The large left atrial mass was firmly adhered to the endocardium at the level of the anterior leaflet of the mitral valve and the interatrial septum. The mass had a grey and whitish appearance with some bluish necrotic patches, (Figure 1B, 2B, 3B).
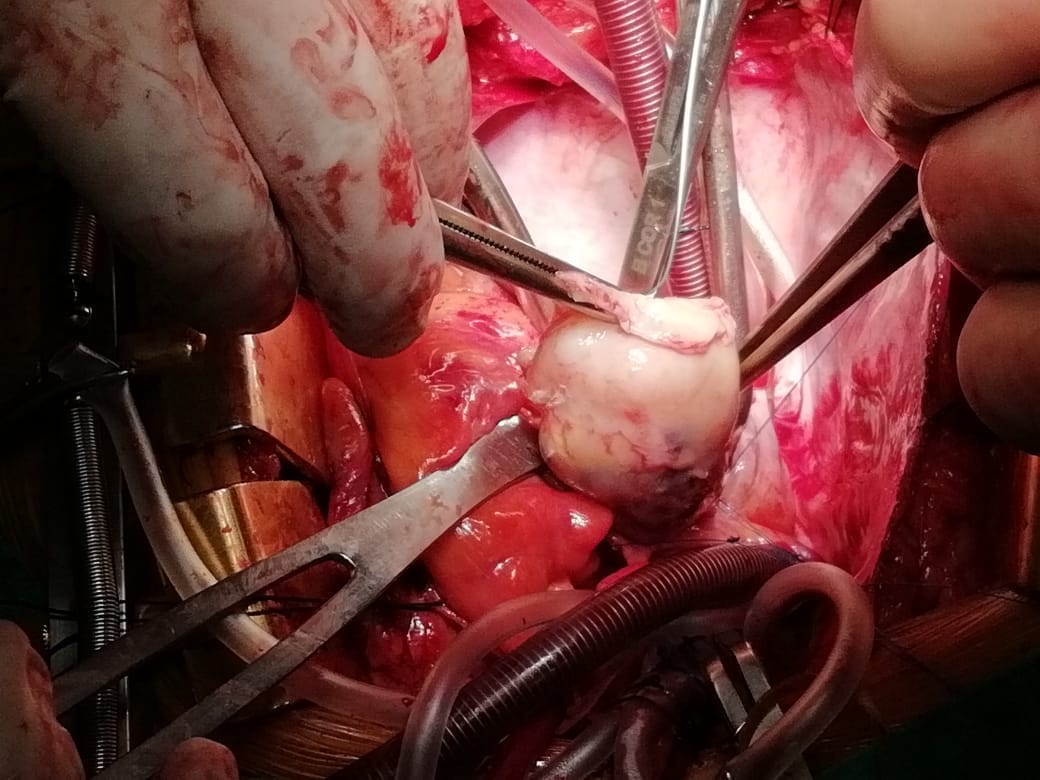
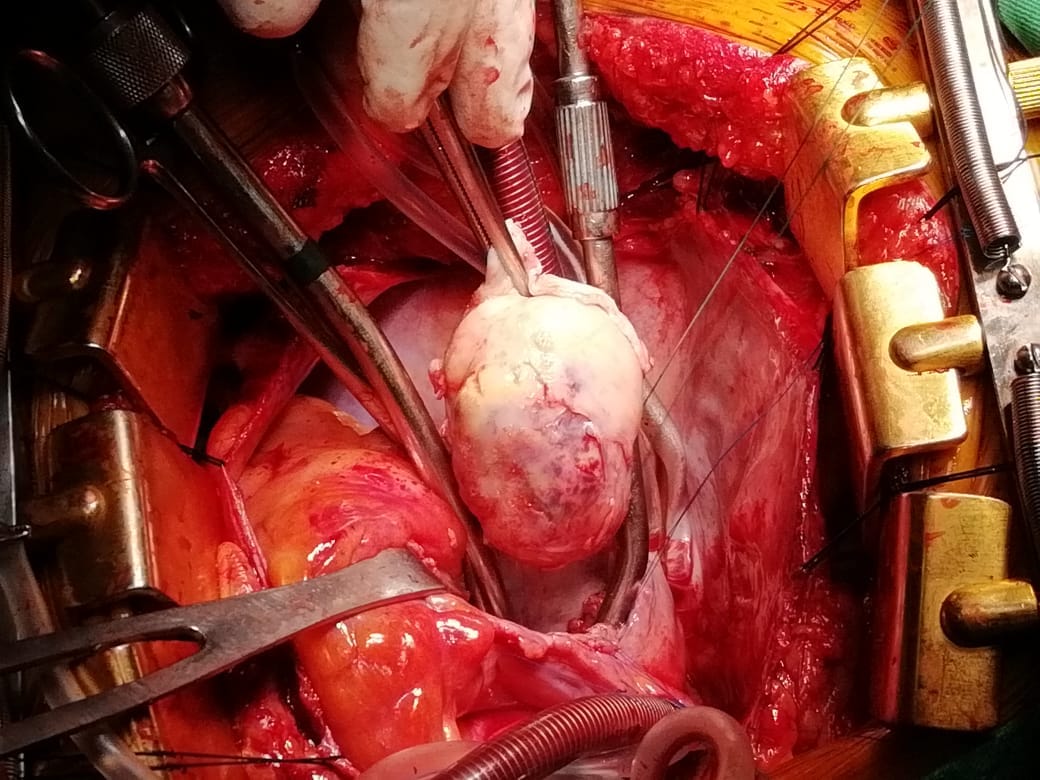
The patient had a complicated postoperative course in the Intensive Care Unit (ICU) and needed inotropic support and vasoactive agents. A postop echocardiogram indicated appropriate left ventricle systolic function, nonetheless, the patient persisted in a hypotensive status that caused refractory shock and ultimately provoked severe organ dysfunction that led to the patient’s death.
Discussion
Primary heart sarcomas are extremely rare malignant neoplasms derived from mesenchymal cells, (1) with an incidence ranging from 0.001% to 0.28% at autopsy.
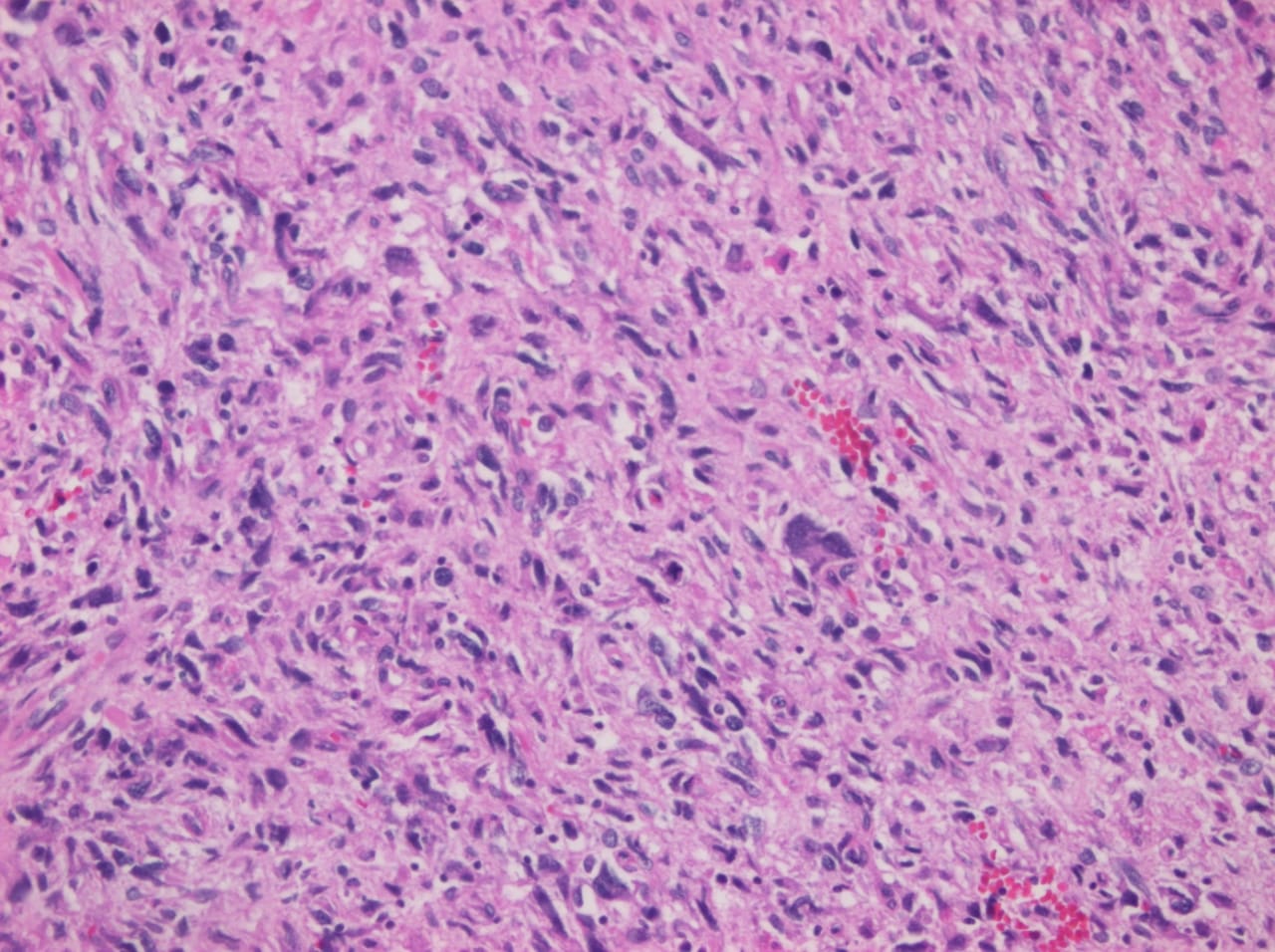
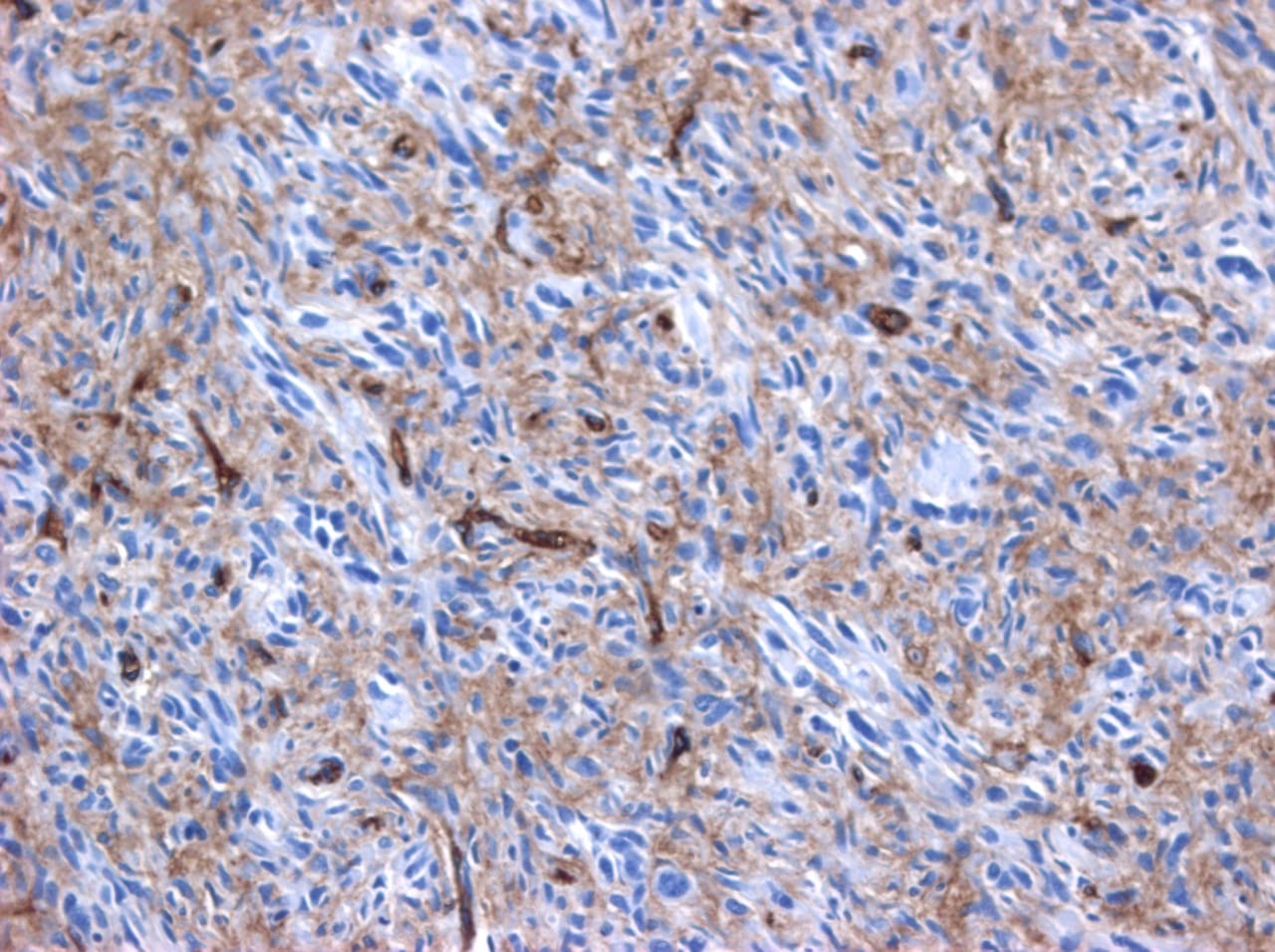
Cardiac angiosarcomas (CA) account for one-third of all primary heart sarcomas (4) and usually develop as gray-brown masses with hemorrhagic patches in the right atrium of male patients. The tumors are filled with vascular channels and their cells are positive for CD34 and factor VIII. (5) Left-sided cardiac angiosarcoma can cause heart failure early in the disease process, but the tumors tend to be more circumscribed, less infiltrative, and associated with better overall survival. (6, 7) Most patients are asymptomatic early in their disease, (2) making the diagnosis even more difficult and worsening its already poor prognosis. (1) The preference of cardiac angiosarcomas for the right heart often leads to a presentation with right-sided congestive heart failure. (2) At later stages, symptoms depend on the structures compromised and range from mild dyspnea on exertion to cardiogenic shock. (8) Cardiac angiosarcomas tend to have a notable intracavitary element, and in some cases may intermittently compromise a cardiac valve, thereby simulating a stenosis or regurgitation. (2, 7)
Our patient presented with acute cardiac failure, pulmonary edema and severe valve dysfunction due to a mass in the left atrium. The tumor had a vascular supply and showed positivity for CD34.
Most patients with cardiac angiosarcoma have metastases, typically to the lung, at diagnosis. (1) Several decades ago, cardiac angiosarcoma was mainly diagnosed postmortem. (1) Now, it can be suspected when cardiomegaly or pleural effusions are seen on chest x-rays (8). Echocardiography is the most useful diagnostic tool, (2) however, CT and MRI can provide useful information on tumor size, invasion and localization. (2, 9) This imaging combination generally provides an excellent anatomic description for preoperative planning. (1, 9)
In our patient, progressive dyspnea was the main symptom and after a prompt evaluation an intracardiac mass was identified as the cause of severe cardiac dysfunction. Because of this finding and the clinical condition of the patient, surgery was planned.
Complete resection of the tumor is the treatment of choice, and is the only therapy currently seen to influence survival. (8) But because of the highly aggressive behavior and a high incidence of systemic metastases with cardiac angiosarcomas, a complete surgical resection is often hampered. (1) Cardiac angiosarcoma carries a grim prognosis as these tumors are universally fatal with a mean survival time of several months after initial presentation even after successful surgery. (2) Chemotherapy is recommended after surgery, even when clear surgical margins are obtained because of the high probability of missed microscopic disease. (1, 2)
High clinical suspicion together with an appropriate history, a thorough physical examination, and precise complementary tests are vital for timely diagnosis and proper treatment.
Authors and Affiliations
Santiago A. Endara: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Gerardo A. Dávalos: Department of General Surgery, Division of Cardiothoracic Surgery, Hospital Metropolitano, Quito, Ecuador, MD
Patricia M. Pontón: Hospital Metropolitano, Quito, Ecuador. Department of Internal Medicine Division of Pathology, MD
Gabriel A. Molina: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY4 General Surgery Resident, MD
Daniel L. Mogrovejo: Pontificia Universidad Católica del Ecuador (PUCE), Quito, Ecuador. PGY1 General Surgery Resident, MD
Corresponding Author Info:
Santiago A. Endara, Hospital Metropolitano, Av. Mariana de Jesus Oe 7/47 y Conclina, Edificio Diagnostico 2000 tercer piso 3/3, Quito, Ecuador, + 593 9 98416157
Email: drsantiagoendara@gmail.com