
Alagille Syndrome: Epidemiology and Management of a Rare Genetic Disease





Alisha Mavis, MD
Clinical Assistant Professor, Department of Pediatrics
Wake Forest University School of Medicine;
Physician, Department of Pediatric Gastroenterology, Hepatology, and Nutrition
Medical Center
Charlotte, North Carolina
Alisha Mavis, MD, has disclosed the following relevant financial relationships: Serve(d) as a director, officer, partner, employee, advisor, consultant, or trustee for: Ipsen; Mirum; Alexion Serve(d) as a member of a speakers bureau for: Ipsen; Mirum; Alexion
Alagille syndrome (ALGS) is a rare, genetically inherited multisystem disorder that typically presents in early childhood.1 The condition is attributed to pathogenic variants in the Notch Homolog 2 (NOTCH2) and jagged canonical Notch ligand 1 (JAG1) genes.1,2 The incidence of ALGS is estimated to be between 1 in 30,000 to 1 in 1,000,000 individuals.1
This condition is characterized by a range of symptoms and anomalies, most notably cholestasis, which can lead to severe liver disease.1 These anomalies can include renal anomalies, cardiac abnormalities, vascular malformations, bone deformities, eye irregularities, and developmental delays.1,3 Genetic testing and diagnostic imaging are key in diagnosis.1 Treatment includes medication to address symptoms─especially pruritus─and liver transplant is not uncommon in these patients.2
The Global Alagille Alliance (GALA) Study comprises more than 100 physicians, surgeons, scientists, and research coordinators from 32 countries around the world. This study aims to produce several significant findings regarding ALGS that contribute to a better understanding of the condition and help improve clinical decision-making and patient care.3,4
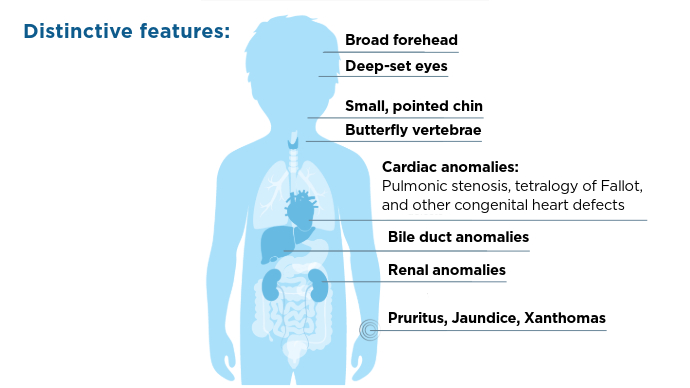 Symptoms and Affected Systems1,3,5
Symptoms and Affected Systems1,3,5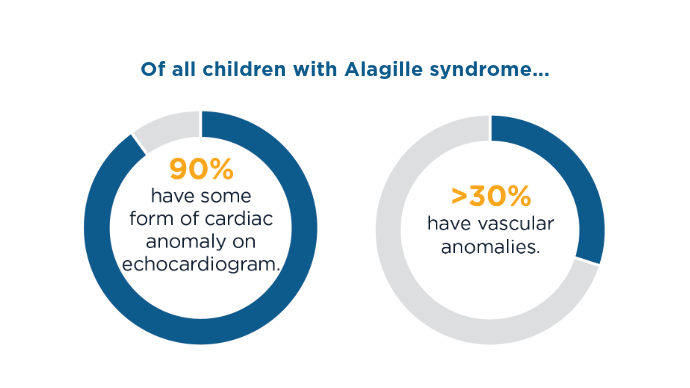 Symptoms and Affected Systems1,3,5
Symptoms and Affected Systems1,3,5 Symptoms and Affected Systems1,3,5
Symptoms and Affected Systems1,3,5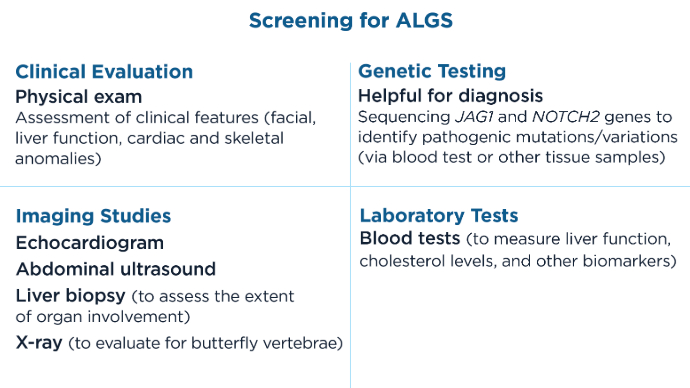 Key Components for Diagnosis and Treatment 1,2,5,6
Key Components for Diagnosis and Treatment 1,2,5,6 Key Components for Diagnosis and Treatment 1,2,5,6
Key Components for Diagnosis and Treatment 1,2,5,6
Managing and treating ALGS involves a multidisciplinary approach to address the various organ systems affected by the condition. The GALA Study: Significant Findings6-9
The GALA Study: Significant Findings6-9- Symptoms and Affected Systems1,3,5
- Symptoms and Affected Systems1,3,5
- Symptoms and Affected Systems1,3,5
- Key Components for Diagnosis and Treatment 1,2,5,6
- Key Components for Diagnosis and Treatment 1,2,5,6
Managing and treating ALGS involves a multidisciplinary approach to address the various organ systems affected by the condition. - The GALA Study: Significant Findings6-9
- Symptoms and Affected Systems1,3,5
- Symptoms and Affected Systems1,3,5
- Symptoms and Affected Systems1,3,5
- Key Components for Diagnosis and Treatment 1,2,5,6
- Key Components for Diagnosis and Treatment 1,2,5,6
Managing and treating ALGS involves a multidisciplinary approach to address the various organ systems affected by the condition. - The GALA Study: Significant Findings6-9