Misleading Diagnosis of Idiopathic Pulmonary Fibrosis: A Clinical Concern





Sjogren syndrome (SS) is a chronic inflammatory autoimmune disorder characterized by lymphocytic infiltration of lacrimal and salivary glands causing sicca syndrome.¹ The disease can extend beyond the exocrine glands, and systemic manifestations, including vasculitis, lung, renal or neurologic involvement, can occur.² Lung disease associated with SS is more commonly seen in women aged ≥ 60 years. The most common symptoms include dry cough, chest pain, and dyspnea on exertion. Sjogren syndrome also may produce several respiratory complications, including bronchial hyperresponsiveness, bronchiolitis, bronchiectasis, pulmonary infections, pulmonary amyloidosis, pulmonary embolism, pulmonary hypertension, lymphomas, and interstitial lung diseases (ILD).² Although ILD typically occurs 5 to 10 years after the onset of SS, lung disease can precede SS.
Pulmonary involvement is associated with systemic manifestations, hypergammaglobulinemia, and anti-SSA and anti-SSB antibodies.² Laboratory tests that confirm a diagnosis of SS include antinuclear antibody (ANA), anti-Ro/SSA, and anti-La/SSB antibodies.² Pulmonary function test (PFT) results appear to reflect impairment of either the lung (restrictive syndrome) or airways (obstructive syndrome).² Imaging abnormalities may include ground-glass attenuation, subpleural small nodules, nonseptal linear opacities, interlobular septal thickening, bronchiectasis, and cysts.³ Therefore, many ILD cases show similar imaging and pathologic findings; nevertheless, they have identifiable etiology that are not idiopathic.
Case Presentation
A 67-year-old man with a medical history of hypertension, peripheral vascular disease, and keratoconjunctivitis sicca (treated with eye drops) developed progressive shortness of breath, dyspnea on exertion, and weight loss (40 pounds) over the course of 6 months. A pulmonary function test showed a restrictive abnormality with decreased diffusing capacity of the lungs for carbon monoxide (eFigure available online at www.fedprac.com). A chest computed tomography (CT) scan showed the presence of significant thickening of the interlobular septi that was more pronounced in the subpleural regions of the lungs and lower lobes, which was consistent with usual interstitial pneumonia. A chest X-ray conducted 4 months prior showed no significant acute cardiopulmonary abnormalities (Figure 1). An open lung wedge biopsy revealed chronic organizing pneumonia with mild interstitial chronic inflammation, smooth muscle hypertrophy, and honeycomb changes consistent with usual interstitial pneumonia. 
The patient had been diagnosed with idiopathic pulmonary fibrosis (IPF) by a private physician and started pirfenidone and oxygen therapy. Three months later the patient presented to the VA Caribbean Healthcare System in San Juan Puerto Rico when he developed an exacerbation of IPF. The patient reported having fever, chills, dry cough, night sweats, and marked shortness of breath. He was found hypoxemic (partial pressure of O2 was 50 mm Hg) and required a venturi mask set to 50% fractioned of inspired O2 to maintain a peripheral oxygen saturation around 90%. A chest X-ray showed decreased lung volume with bilateral interstitial and alveolar disease (Figure 2). Leukocytosis was present at 17×10-3/µl. The chest CT scan showed interval worsening of diffuse ground-glass airspace opacities and worsening of interstitial opacities; there 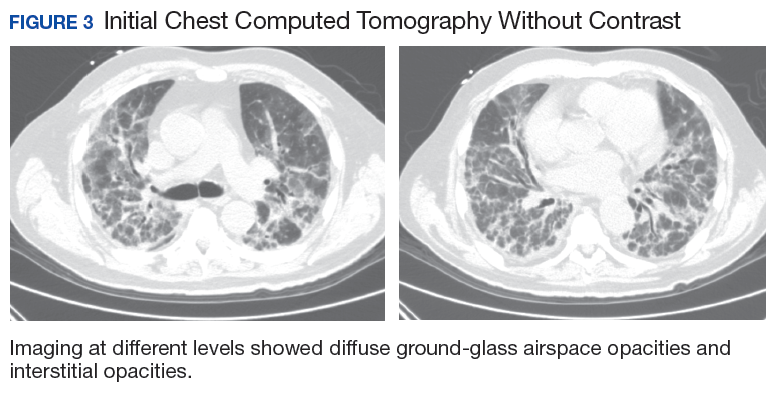
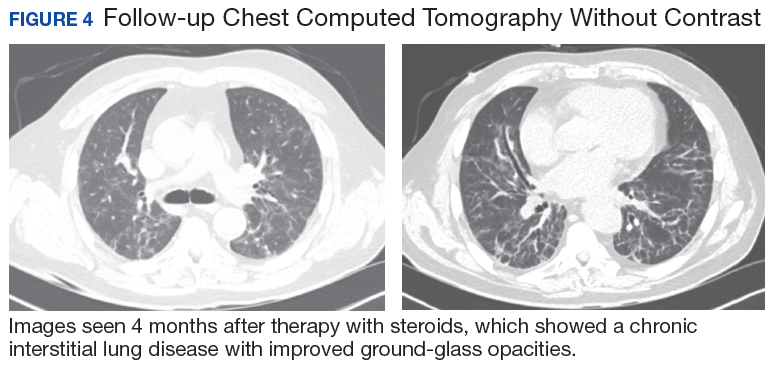
After careful clinical assessment (+ dry eyes) and radiographic pattern evaluation (diffuse bilateral interstitial and ground-glass opacities), the clinical diagnosis of IPF was queried after the patient’s rheumatologic workup came back positive for ANA and anti-Ro/SSA tests. Since the etiology of ILD was secondary to SS, pirfenidone was discontinued, and the patient was started on steroid therapy with subsequent marked clinical improvement. Parotid biopsy revealed the presence of inflammatory cells supporting the diagnosis of ILD associated to SS. The patient was discharged home on a tapering dose of steroids. Four months after therapy with steroids, a follow-up chest CT scan without contrast showed a chronic ILD with improved ground-glass opacities (Figure 4). The patient currently is in good health without oxygen supplementation.
Discussion
Diagnosis of SS is challenging, since it may mimic other conditions such as IPF. The most common type of SS-associated ILD is nonspecific interstitial pneumonia (NSIP), although usual interstitial pneumonia (UIP) can be visualized, as in this case study. Usual interstitial pneumonia